
ABSTRACT
The mechanisms leading to brain tumor formation are poorly understood. Using Ptch1+/− mice as a medulloblastoma model, sequential mutations were found to shape tumor evolution. Initially, medulloblastoma preneoplastic lesions display loss of heterozygosity of the Ptch1 wild-type allele, an event associated with cell senescence in preneoplasia. Subsequently, p53 mutations lead to senescence evasion and progression from preneoplasia to medulloblastoma. These findings are consistent with a model where high levels of Hedgehog signaling caused by the loss of the tumor suppressor Ptch1 lead to oncogene-induced senescence and drive p53 mutations. Thus, cell senescence is an important characteristic of a subset of SHH medulloblastoma and might explain the acquisition of somatic TP53 mutations in human medulloblastoma. This mode of medulloblastoma formation contrasts with the one characterizing Li-Fraumeni patients with medulloblastoma, where TP53 germ-line mutations cause chromothriptic genomic instability and lead to mutations in Hedgehog signaling genes, which drive medulloblastoma growth. Here we discuss in detail these 2 alternative mechanisms leading to medulloblastoma tumorigenesis.
Introduction
Medulloblastoma is the most common brain tumor in children. According to integrated genomics studies, medulloblastomas can be classified in 4 different molecular subtypes: WNT, SHH, Group 3 and Group 4.Citation1 Each of these molecular subtypes has specific underlying molecular features, gene expression, demographic characteristics and prognosis.Citation2 Sonic Hedgehog (SHH) medulloblastomas are characterized by activation of the Hedgehog (Hh) signaling pathway and are often driven by mutations in Hh pathway components.Citation3 Here we focus on SHH medulloblastoma and discuss how cell senescence shapes the natural history and molecular evolution of this childhood cancer.
TP53 mutations and medulloblastoma
For many years, it was thought that TP53 mutations were infrequent in medulloblastomasCitation4,5 and that P53 signaling was dispensable for medulloblastoma tumor suppression. However, recent studies reported that TP53 mutations are frequent in human WNT (with a rate of 16%) and SHH primary medulloblastomas (with a rate of 13% to 21%, depending on the study)Citation3,6,7 and are indicators of poor prognosis exclusively in SHH medulloblastoma.Citation7 Moreover, TP53 mutations have recently been identified as a key event in the pathogenesis of medulloblastoma recurrence.Citation8,9 Although the specific roles of P53 in medulloblastoma are still largely unknown,Citation10 the presence of recurrent TP53 mutations suggests that P53 signaling plays an important role in driving medulloblastoma tumorigenesis.
Human genomic studies also established that TP53 mutations co-exist with mutations or amplifications of Hh signaling components such as SHH, SMO, SUFU, GLI2 and MYCN.Citation3 These studies, however, do not establish the sequential order of genetic events that lead to medulloblastoma formation and how these mutations correlate with presumptive histopathological stages of medulloblastoma. Nevertheless, patients with Li-Fraumeni syndrome, caused by germ-line mutations in TP53, offer an opportunity to study how medulloblastomas arise at the genetic level. Li-Fraumeni patients are cancer-prone and sometimes develop medulloblastoma;Citation11 therefore, at least for this subset of patients, TP53 mutations are the first genetic event leading to medulloblastoma formation (). Notably, most Li-Fraumeni medulloblastomas belong to the SHH subgroup, indicating that TP53 mutations specifically predispose to SHH medulloblastoma.Citation12 Because cerebellum granule cell precursors (GCPs) are the cells of origin of SHH medulloblastoma,Citation13-15 this indicates that GCPs are highly susceptible to transformation in absence of P53. Moreover, these SHH medulloblastomas seem to be the result of chromothripsis, a massive genome rearrangement caused by chromosomal shattering likely occurring in a single event.Citation16 These chromothriptic events likely lead to mutations in components of Hh signaling, such as GLI2, BOC and MYCN amplifications.Citation12 Since Hh signaling is the most important mitogenic pathway for GCPs,Citation17 it is therefore expected that acquisition of Hh pathway mutations efficiently causes SHH medulloblastoma in Li-Fraumeni patients.
Figure 1. Two alternative mechanisms of medulloblastoma formation. (A) In Li-Fraumeni patients with germ-line TP53 mutations, cerebellum GCPs experience chromosomal instability. Chromothriptic events lead to massive chromosomal rearrangements and high levels of amplification in Hh signaling genes such as GLI2 and MYCN. Acquisition of these Hh signaling mutations leads to medulloblastoma growth. (B) In Ptch1+/− mice, loss of heterozygosity of the Ptch1 wild-type allele leads to the formation of preneoplasia. Preneoplastic lesions display high levels of cell senescence. Spontaneous p53 mutations or p16ink4a inactivation leads to senescence evasion and progression to advanced medulloblastoma. Proliferation and senescence levels during medulloblastoma formation are indicated.
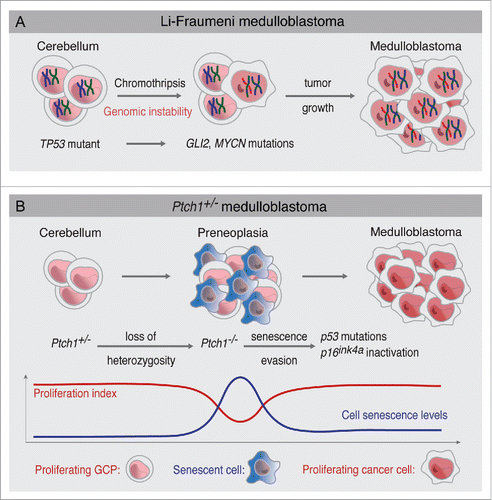
Although p53 knockout mice or mouse models of Li-Fraumeni syndrome do not develop medulloblastoma,Citation18,19 elegant studies have demonstrated that the inactivation of p53 together with other DNA repair factors such as as Xrcc4, Ligase IV, Xrcc2 and Brca2 leads to Shh medulloblastoma.Citation20,21 Interestingly, those medulloblastomas also harbored spontaneous mutations in Hh signaling components such as Ptch1, Mycn and Gli2.Citation20 This result not only highlights how important it is to maintain genomic stability to prevent SHH medulloblastoma formation, but it also indicates that Hh signaling activation seems to be necessary for medulloblastoma formation even when p53 and DNA repair mechanisms are absent. In summary, germ-line P53 mutations in both mouse and human cause genomic instability and lead to mutations in Hh signaling components, conducing to medulloblastoma formation (). This is an interesting paradigm showing that Hh signaling mutations happen subsequent to TP53 mutations in Li-Fraumeni syndrome.
Half of the SHH medulloblastomas with TP53 mutations have a germ-line (Li-Fraumeni) origin and are potentially explained by the mechanism described above.Citation3,12 However, it is not known how the other SHH medulloblastoma cases (including the ones with somatic TP53 mutations and the ones without TP53 mutations) develop and the temporal order in which they acquire their mutations. Another unresolved, yet related, question is whether advanced medulloblastomas arise in a step-wise manner from subclinical precancerous lesions. For many epithelial cancers, the availability of preneoplastic lesions allowed the establishment of tumor progression models with sequential histopathological stages and the molecular changes that characterize them.Citation22 However, the problem for understanding brain tumor development lies in the inability to detect and obtain precancerous lesions; therefore, genome sequencing of advanced brain tumors only offers a snapshot of the mutations present in advanced tumors but does not show the order in which mutations are acquired during tumor progression.
To address this question, we used Ptch1+/− heterozygous mice, a well-established model of Shh medulloblastoma (). Ptch1+/− mice develop preneoplastic lesions with high frequency, but only a fraction of those animals develop advanced medulloblastoma.Citation13,23 Therefore, we hypothesized that an unidentified tumor suppressive mechanism might restrain the progression of medulloblastoma preneoplasia into advanced tumors. We found that apoptosis levels are the same between preneoplastic lesions and advanced medulloblastoma, eliminating apoptosis as an essential tumor suppressor in this model of Shh medulloblastoma. Surprisingly, when we looked at cell senescence, we found that while preneoplastic lesions display high numbers of p21Cip1 and p16Ink4a positive cells (which are effectors and markers of cell senescence and cell cycle arrestCitation24-26), advanced medulloblastomas have very low levels of senescence.Citation27 These high levels of senescence are paralleled by lower levels of proliferation and correlate with loss of heterozygosity (LOH) of the Ptch1 wild-type allele in preneoplastic lesions (), suggesting that high levels of Hh signaling may contribute to cell senescence. Using laser capture microdissection, we found that one-third of all advanced medulloblastomas acquired p53 mutations that were not present in preneoplastic lesions, supporting the notion that p53 mutations allow senescence evasion and medulloblastoma progression (). Moreover, we found that engineered p53 mutations prevent cellular senescence and accelerate medulloblastoma formation, showing that p53 mutations lead to senescence evasion.Citation27 Further supporting the idea that senescence evasion is necessary for medulloblastoma progression, advanced tumors without p53 mutations display low expression of p16Ink4a due to promoter methylation. In summary, we found that, contrary to Li-Fraumeni syndrome (where TP53 germ-line mutations lead to Hh signaling mutations), Hh signaling hyperactivity leads to cell senescence in preneoplastic lesions and creates selection pressure for the inactivation of p53 or p16Ink4a, which allows the progression from preneoplasia to advanced medulloblastoma ().
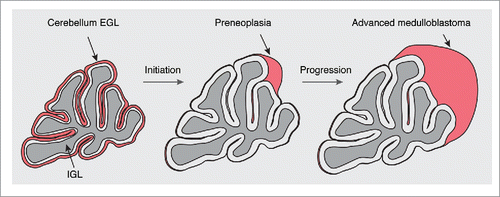
Figure 2. Medulloblastoma formation in Ptch1+/− mice. During postnatal development, granule cell precursors (GCPs) of the cerebellum, the cells of origin of Shh medulloblastoma, are located in the external granule-cell layer (EGL). After their proliferation, most GCPs differentiate, populate the internal granule-cell layer (IGL), and disappear from the EGL after the second postnatal week in the mouse. LOH of the Ptch1 wild-type allele causes a clonal expansion and leads to the formation of preneoplasia. While most preneoplastic lesions disappear, some of them progress to advanced medulloblastoma. See also .
Importantly, the finding of spontaneous p53 mutations is not limited to the Ptch1+/− model, as we also found spontaneous p53 mutations in tumors from Olig1-Gnas mice, another model of Shh medulloblastoma.Citation28 This suggests that evasion of senescence could be a hallmark of Shh medulloblastoma.
Hh and p53 signaling in medulloblastoma
Since p53 is inactivated during the formation of Shh medulloblastoma (), this may highlight some possible interactions between p53 and Hh signaling in the brain. Some studies have explored this relationship using a variety of approaches. Shh, a protein secreted by Purkinje cells of the cerebellum, is the most potent mitogen for GCPs.Citation17,29,30 Shh also promotes proliferation of neural progenitors of the adult hippocampus.Citation31 In contrast to these proliferative effects of Shh, p53 activity supresses proliferation and self-renewal of adult neural stem cells of the subventricular zone,Citation32 and this effect might be mediated by p21Cip1.Citation32,33 This may explain why p53 activity is downregulated during neurogenesisCitation34 to allow cell proliferation and brain formation. Interestingly, Gli activity has been shown to downregulate p53 protein levels in cell lines.Citation35 Specifically, it has been proposed that high levels of Hh signaling in cell lines caused by expression of constitutively active Smo mutants or overexpression of Gli1 and Gli2 leads to an Mdm2-dependent degradation of p53.Citation36 Consistently, low levels of Mdm2 in vivo increase p53 levels, lead to cerebellar hypoplasia and reduce medulloblastoma development in Ptch1+/− mice,Citation37 showing that p53 signaling negatively controls cerebellum growth and implying that p53 signaling is important for medulloblastoma tumor suppression. Similarly, the proto-oncogene PPM1D, a negative regulator of p53, is overexpressed in medulloblastomasCitation38 and increases medulloblastoma formation in mice when overexpressed together with Shh.Citation39 Together, these findings provide strong evidence that Hh signaling leads to a functional inactivation of p53 signaling that allows GCP proliferation and, in some instances, medulloblastoma formation. However, the presence of somatic p53 mutations in Ptch1+/− medulloblastomaCitation27 demonstrates that the ability of Shh signaling to functionally suppress p53 signaling is not always sufficient to inactivate p53 activity in a tumorigenic context.
For many years, it has been known that p53 deletion accelerates medulloblastoma formation and increases medulloblastoma incidence in Ptch1+/− mice;Citation40 however, the mechanism responsible for this was never investigated. The fact that p53 mutations, which are acquired spontaneously during medulloblastoma formation, lead to senescence evasion provides an explanation for the presence of somatic TP53 mutations in human SHH medulloblastoma.Citation27
Cell senescence as a driver of p53 inactivation in medulloblastoma
Gli1 or Smo overexpression in cell lines causes high levels of oncogenic stress.Citation35,36 Recently, we also found that the ligand Shh induces DNA damage in GCPs, an effect that requires the presence of the Hh receptor Boc and CyclinD1.Citation23 These results support the hypothesis that GCPs are sensitive to replicative stressCitation41 and that hedgehog signaling likely causes replication stress. It has been demonstrated that oncogene activation leads to oncogene-induced cell senescence (OIS), a tumor-suppressive mechanism that prevents transformation of premalignant lesions into tumors.Citation42 Moreover, oncogene-induced DNA damage seems to be required for OIS.Citation43,44 In light of this, the fact that we found cell senescence and Ptch1 LOH in medulloblastoma preneoplastic lesionsCitation27 suggests that high levels of hedgehog signaling in preneoplasia likely cause high levels of oncogenic stress and this leads to cell senescence. Therefore, we propose that high levels of hedgehog signaling shape the molecular evolution of medulloblastoma by leading to OIS and creating selection pressure to inactivate p53 in order to evade OIS.
In addition to oncogenes, loss of tumor suppressor genes has been shown to cause OIS.Citation45 For example, in the context of prostate cancer, Pten inactivation causes senescence and leads to the acquisition of p53 mutations.Citation46 Neurofibromin (NF1) loss also leads to Ras-mediated induction of cell senescence.Citation47 Since we observed a strong association between Ptch1 loss and cell senescence in medulloblastoma preneoplasia,Citation27 Ptch1 seems to be a new tumor suppressor whose absence may be capable of causing OIS.
TP53 mutations are also present in WNT medulloblastoma.Citation7 However, the molecular mechanism leading to TP53 mutations has never been investigated in WNT medulloblastoma. Interesting work has shown that, at least in cell lines, overexpression of the downstream Wnt effector ß-catenin leads to p53 stabilization and activation.Citation48 Wnt activation in colorectal cancer has been associated with decreased levels of proliferationCitation49 and accumulation of p53 protein in early tumor stages,Citation50 suggesting that Wnt signaling may lead to cell senescence in precancerous lesions and this could be the cause of the acquisition of TP53 mutations at late stages of colorectal cancer. Additionally, it was shown that ß-catenin overexpression causes OIS in thymocytes.Citation51 We thus speculate that Wnt activation in the brainstem may also lead to changes compatible with cell senescence and this may be an explanation for the presence of TP53 mutations in advanced WNT medulloblastomas. This is supported by the fact that expression of an active ß-catenin mutant in the lower rhombic lip of mice leads to the formation of hyperplastic lesions that only progress to advanced medulloblastomas when p53 is also deleted.Citation52
High-grade astrocytomas frequently harbor TP53 mutations.Citation53 It has been shown that low-grade astrocytoma lesions display a DNA damage responseCitation54,55 and that loss of components of the Atm-Chk2-p53 pathway accelerates astrocytoma development.Citation55 Although cell senescence was not interrogated in those reports, another study showed that pilocytic astrocytomas (PA), the most benign type of astrocytomas, display MAPK activation and OIS;Citation56 interestingly, PA are indolent tumors that display long periods of growth arrest, rarely become high-grade astrocytomas, and do not display TP53 mutations. This is in agreement with the idea that PA are long-term senescent astrocytic lesions that do not progress.Citation56
Abbreviations
| EGL | = | External Granule-cell Layer |
| GCPs | = | Granule Cell Precursors |
| Hh | = | Hedgehog |
| IGL | = | Internal Granule-cell Layer |
| OIS | = | Oncogene-induced senescence |
| Shh | = | Sonic hedgehog |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Work done in the Charron lab was supported by grants from the Canadian Institutes of Health Research (CIHR), the Fonds de Recherche du Québec-Santé (FRQS), and the Canada Foundation for Innovation (CFI). LT-O is recipient of the Caldas fellowship (Colciencias). SMS is recipient of the McGill James O. and Maria Meadows fellowship and IRCM Challenge-Guépards et Gazelles gourmands scholarship. FC holds the Canada Research Chair in Developmental Neurobiology.
References
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012; 123:465-72; PMID:22134537; http://dx.doi.org/10.1007/s00401-011-0922-z
- Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 2012; 123:473-84; PMID:22358457; http://dx.doi.org/10.1007/s00401-012-0958-8
- Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014; 25:393-405; PMID:24651015; http://dx.doi.org/10.1016/j.ccr.2014.02.004
- Adesina AM, Nalbantoglu J, Cavenee WK. p53 gene mutation and mdm2 gene amplification are uncommon in medulloblastoma. Cancer Res 1994; 54:5649-51; PMID:7923211
- Saylors RL, 3rd, Sidransky D, Friedman HS, Bigner SH, Bigner DD, Vogelstein B, Brodeur GM. Infrequent p53 gene mutations in medulloblastomas. Cancer Res 1991; 51:4721-3; PMID:1873817
- Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2011; 331:435-9; PMID:21163964; http://dx.doi.org/10.1126/science.1198056
- Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC, Castelo-Branco P, Baskin B, Ray PN, Bouffet E, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 2013; 31:2927-35; PMID:23835706; http://dx.doi.org/10.1200/JCO.2012.48.5052
- Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, Remke M, Cavalli FM, Ramaswamy V, Lindsay PE, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016; 529:351-7; PMID:26760213; http://dx.doi.org/10.1038/nature16478
- Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, Boult JK, Williamson D, Ahmad Z, Hallsworth A, et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015; 27:72-84; PMID:25533335; http://dx.doi.org/10.1016/j.ccell.2014.11.002
- Ramaswamy V, Nor C, Taylor MD. p53 and Meduloblastoma. Cold Spring Harb Perspect Med 2015; 6:1-9; PMID:26684332.
- Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 1997; 150:1-13; PMID:9006316
- Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012; 148:59-71; PMID:22265402; http://dx.doi.org/10.1016/j.cell.2011.12.013
- Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, Lin SM, Wechsler-Reya RJ. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development 2005; 132:2425-39; PMID:15843415; http://dx.doi.org/10.1242/dev.01793
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer cell 2008; 14:123-34; PMID:18691547; http://dx.doi.org/10.1016/j.ccr.2008.07.005
- Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 2008; 14:135-45; PMID:18691548; http://dx.doi.org/10.1016/j.ccr.2008.07.003
- Jones MJ, Jallepalli PV. Chromothripsis: chromosomes in crisis. Dev Cell 2012; 23:908-17; PMID:23153487; http://dx.doi.org/10.1016/j.devcel.2012.10.010
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999; 22:103-14; PMID:10027293; http://dx.doi.org/10.1016/S0896-6273(00)80682-0
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994; 4:1-7; PMID:7922305; http://dx.doi.org/10.1016/S0960-9822(00)00002-6
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119:847-60; PMID:15607980; http://dx.doi.org/10.1016/j.cell.2004.11.004
- Frappart PO, Lee Y, Russell HR, Chalhoub N, Wang YD, Orii KE, Zhao J, Kondo N, Baker SJ, McKinnon PJ. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc Natl Acad Sci U S A 2009; 106:1880-5; PMID:19164512; http://dx.doi.org/10.1073/pnas.0806882106
- Yan CT, Kaushal D, Murphy M, Zhang Y, Datta A, Chen C, Monroe B, Mostoslavsky G, Coakley K, Gao Y, et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc Natl Acad Sci U S A 2006; 103:7378-83; PMID:16670198; http://dx.doi.org/10.1073/pnas.0601938103
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61:759-67; PMID:2188735; http://dx.doi.org/10.1016/0092-8674(90)90186-I
- Mille F, Tamayo-Orrego L, Levesque M, Remke M, Korshunov A, Cardin J, Bouchard N, Izzi L, Kool M, Northcott PA, et al. The Shh receptor Boc promotes progression of early medulloblastoma to advanced tumors. Dev Cell 2014; 31:34-47; PMID:25263791; http://dx.doi.org/10.1016/j.devcel.2014.08.010
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729-40; PMID:17667954; http://dx.doi.org/10.1038/nrm2233
- Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur J Biochem 2001; 268:2784-91; PMID:11358493; http://dx.doi.org/10.1046/j.1432-1327.2001.02228.x
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22:4212-22; PMID:12912919; http://dx.doi.org/10.1093/emboj/cdg417
- Tamayo-Orrego L, Wu CL, Bouchard N, Khedher A, Swikert SM, Remke M, Skowron P, Taylor MD, Charron F. Evasion of cell senescence leads to medulloblastoma progression. Cell Rep 2016; 14:2925-37; PMID:26997276; http://dx.doi.org/10.1016/j.celrep.2016.02.061
- He X, Zhang L, Chen Y, Remke M, Shih D, Lu F, Wang H, Deng Y, Yu Y, Xia Y, et al. The G protein α subunit Galphas is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat Med 2014; 20:1035-42; PMID:25150496; http://dx.doi.org/10.1038/nm.3666
- Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 1999; 126:3089-100; PMID:10375501
- Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 1999; 9:445-8; PMID:10226030; http://dx.doi.org/10.1016/S0960-9822(99)80195-X
- Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci 2003; 6:21-7; PMID:12469128; http://dx.doi.org/10.1038/nn983
- Meletis K, Wirta V, Hede SM, Nister M, Lundeberg J, Frisen J. p53 suppresses the self-renewal of adult neural stem cells. Development 2006; 133:363-9; PMID:16368933; http://dx.doi.org/10.1242/dev.02208
- Kippin TE, Martens DJ, van der Kooy D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev 2005; 19:756-67; PMID:15769947; http://dx.doi.org/10.1101/gad.1272305
- Louis JM, McFarland VW, May P, Mora PT. The phosphoprotein p53 is down-regulated post-transcriptionally during embryogenesis in vertebrates. Biochim Biophys Acta 1988; 950:395-402; PMID:3048409; http://dx.doi.org/10.1016/0167-4781(88)90136-4
- Stecca B, Ruiz i Altaba A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J 2009; 28:663-76; PMID:19214186; http://dx.doi.org/10.1038/emboj.2009.16
- Abe Y, Oda-Sato E, Tobiume K, Kawauchi K, Taya Y, Okamoto K, Oren M, Tanaka N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc Natl Acad Sci U S A 2008; 105:4838-43; PMID:18359851; http://dx.doi.org/10.1073/pnas.0712216105
- Malek R, Matta J, Taylor N, Perry ME, Mendrysa SM. The p53 inhibitor MDM2 facilitates Sonic Hedgehog-mediated tumorigenesis and influences cerebellar foliation. PloS One 2011; 6:e17884; PMID:21437245; http://dx.doi.org/10.1371/journal.pone.0017884
- Castellino RC, De Bortoli M, Lu X, Moon SH, Nguyen TA, Shepard MA, Rao PH, Donehower LA, Kim JY. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol 2008; 86:245-56; PMID:17932621; http://dx.doi.org/10.1007/s11060-007-9470-8
- Doucette TA, Yang Y, Pedone C, Kim JY, Dubuc A, Northcott PD, Taylor MD, Fults DW, Rao G. WIP1 enhances tumor formation in a sonic hedgehog-dependent model of medulloblastoma. Neurosurgery 2012; 70:1003-10; discussion 10; PMID:22037313; http://dx.doi.org/10.1227/NEU.0b013e31823e5332
- Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res 2001; 61:513-6; PMID:11212243
- McKinnon PJ. Maintaining genome stability in the nervous system. Nat Neurosci 2013; 16:1523-9; PMID:24165679; http://dx.doi.org/10.1038/nn.3537
- Narita M, Lowe SW. Senescence comes of age. Nat Med 2005; 11:920-2; PMID:16145569; http://dx.doi.org/10.1038/nm0905-920
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638-42; PMID:17136094; http://dx.doi.org/10.1038/nature05327
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633-7; PMID:17136093; http://dx.doi.org/10.1038/nature05268
- d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 2008; 8:512-22; PMID:18574463; http://dx.doi.org/10.1038/nrc2440
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:725-30; PMID:16079851; http://dx.doi.org/10.1038/nature03918
- Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 2006; 10:459-72; PMID:17157787; http://dx.doi.org/10.1016/j.ccr.2006.10.003
- Damalas A, Ben-Ze'ev A, Simcha I, Shtutman M, Leal JF, Zhurinsky J, Geiger B, Oren M. Excess β-catenin promotes accumulation of transcriptionally active p53. EMBO J 1999; 18:3054-63; PMID:10357817; http://dx.doi.org/10.1093/emboj/18.11.3054
- Mahmoud NN, Boolbol SK, Bilinski RT, Martucci C, Chadburn A, Bertagnolli MM. Apc gene mutation is associated with a dominant-negative effect upon intestinal cell migration. Cancer Res 1997; 57:5045-50; PMID:9371501
- Tominaga O, Hamelin R, Trouvat V, Salmon RJ, Lesec G, Thomas G, Remvikos Y. Frequently elevated content of immunochemically defined wild-type p53 protein in colorectal adenomas. Oncogene 1993; 8:2653-8; PMID:8378077
- Xu M, Yu Q, Subrahmanyam R, Difilippantonio MJ, Ried T, Sen JM. Beta-catenin expression results in p53-independent DNA damage and oncogene-induced senescence in prelymphomagenic thymocytes in vivo. Mol Cell Biol 2008; 28:1713-23; PMID:18160717; http://dx.doi.org/10.1128/MCB.01360-07
- Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010; 468:1095-9; PMID:21150899; http://dx.doi.org/10.1038/nature09587
- Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061-8; PMID:18772890; http://dx.doi.org/10.1038/nature07385
- Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H, et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010; 29:5095-102; PMID:20581868; http://dx.doi.org/10.1038/onc.2010.249
- Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 2010; 18:619-29; PMID:21156285; http://dx.doi.org/10.1016/j.ccr.2010.10.034
- Jacob K, Quang-Khuong DA, Jones DT, Witt H, Lambert S, Albrecht S, Witt O, Vezina C, Shirinian M, Faury D, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res 2011; 17:4650-60; PMID:21610151; http://dx.doi.org/10.1158/1078-0432.CCR-11-0127