
Post-translational modifications of target DNA repair proteins may help to regulate the mechanism of DNA damage correction. A good analogy is an automobile tune-up necessary to make a car operate at its optimal capacity (). Although the vehicle may be serviceable, an oil change or spark plug replacement may improve engine performance. In a similar manner, the functionality of the cell’s DNA repair machinery may be boosted by a covalent modification of a target protein responsible for removal of the damaged base to suppress mutation. In a recent work, Piekna-Przybylska et al. assessed biological effects of protein acetylation on DNA repair capacity in human cells.Citation1 The researchers’ experimental approach was based on flow cytometric analysis of human cells transfected with a reporter DNA plasmid harboring a site-specific oxidative base lesion (8-oxoguanine (8-oxoG)) or base-base mismatch that disrupted coding sequence of a green fluorescent marker gene. The reporter plasmid DNA construct contained both the lesion and a single-strand nick, the latter providing a site for DNA repair machinery (e.g., mismatch repair (MMR)) to load onto the substrate and excise the damaged strand so it can be replaced with normal sequence. The DNA repair assay monitored correction of the abnormal DNA base in cells defective in MMR and/or a major acetyltransferase known as p300. Alternatively, MMR-proficient or deficient cells were pharmacologically modulated with deacetylase or acetyltransferase inhibitors to assess the importance of acetylation status on DNA repair capacity.
Figure 1. Fine-tuning DNA repair by acetylation. Just as an automobile tune-up may help the engine run more smoothly, post-translational modification of DNA repair machinery by acetylation may enhance its functionality, as demonstrated in the recent Cell Cycle paper by Piekna-Przybylska et al.Citation1 Artwork is courtesy of Tom Wynn, Visual Media, NIA-NIH.
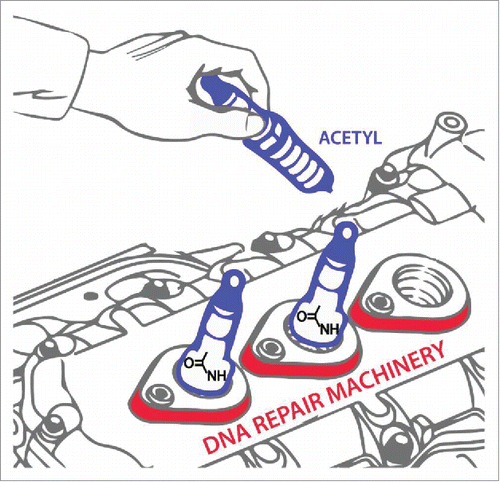
Pharmacological inhibition of deacetylase activity was observed to up-regulate nick-directed DNA repair of a mismatch in the cell-based assay.Citation1 Consistent with this finding, cellular exposure to an acetyltransferase inhibitor decreased repair of mismatches or nicks, even in cells genetically defective in MLH1, a key component of the MMR machinery. Presumably, acetylation of a human nuclease involved in replication intermediate processing (e.g., DNA2 implicated in Okazaki fragment maturationCitation2) increases repair of duplex DNA containing a nick or mismatch; however, the precise mechanism remains to be determined. Alternatively, acetylation (and other post-translational modifications) of a MMR helicase or DNA polymerase might alter its catalytic DNA efficiency, which in turn affects strand displacement synthesis. Interestingly, pharmacological modulation of acetylation did not affect 8-oxoG correction, suggesting that post-translational regulatory control of base excision repair (BER), even long patch BER processing of DNA flaps, is distinct from that which operates for mismatch correction.
Because p300 acetyltransferase is known to acetylate non-histone and histone proteins,Citation3 the authors investigated the effect of p300 deficiency on DNA repair and found that cells were significantly compromised in 5’-nick directed mismatch correction irrespective of MLH1 mutational status,Citation1 further suggesting that acetylation promotes excision of the tract containing the mismatched base independent of the classic MMR pathway. However, p300 deficiency exerted no effect on BER of 8-oxoG. This result was unexpected, given a number of in vitro studies showing that BER proteins can be acetylated and that long patch BER in reconstituted cell extract systems was affected by acetylation.Citation4 The existence of other acetyltransferases engaged in the DNA damage response in human cells (CBP, p300, PCAF, Tip60, MOF)Citation5 may negate the effect of p300 deficiency on BER, or the scenario may be more complex. It will be important to assess precisely which acetyltransferase(s) serves as the best bull’s eye for DNA repair modulation.
A significant advance in the current work was that the scientists addressed the importance of acetylation in DNA repair in living human cells, whereas the majority of previous studies examined the effect of acetylation on DNA replication or repair in vitro using reconstituted systems. Although the plasmid reporter system is valuable in that it focuses on acetylation of DNA repair proteins and likely excludes effects of acetylation on chromatin state and therefore lesion access, it will be important to validate the findings of the current work for clinically relevant chromosomal genomic lesions. This may provide a better understanding of how emerging therapies focused on DNA damage response pathways can be fine-tuned. In addition, lagging strand replication errorsCitation6 could be a suitable target for acetylation-based cancer therapy. If acetylation truly is a direct modulator of DNA repair, it will be relevant at the translational level to affect disease-related pathways and clinical features of aging precipitated by genomic instability and DNA repair defects.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by the Intramural Research program of the NIH, National Institute on Aging.
References
- Piekna-Przybylska D, et al. Cell Cycle in press; 2016; 15(11):1506—17; PMID: 27104361; http://dx.doi.org/10.108015384101.2016.1176815
- Balakrishnan L, et al. J Biol Chem 2010; 285:4398–404; PMID:20019387; http://dx.doi.org/10.1074/jbc.M109.086397
- Dancy BM, Cole PA. Chem Rev 2015; 115:2419–52; PMID:25594381; http://dx.doi.org/10.1021/cr500452k
- Carter RJ, Parsons JL. Mol Cell Biol 2016; 36:1426–37; PMID:26976642; http://dx.doi.org/10.1128/MCB.00030-16
- Chatterjee S, et al. Essays Biochem 2012; 52:93–111; PMID:22708566; http://dx.doi.org/10.1042/bse0520093
- Reijns MA, et al. Nature 2015; 518:502–6; PMID:25624100; http://dx.doi.org/10.1038/nature14183