
The generation of cancer-specific gene expression patterns is attributed to aberrant activation of oncogenic transcription factors or repression of tumor suppressors. It is also well established that transcription factors synergize with enzymes that catalyze changes in chromatin structure to elicit their effects in gene activation. Deregulated expression or altered activity of chromatin modifying enzymes play pivotal role in the generation of cancer-specific “epigenomes” (the global array of DNA and histone modifications), which are causatively related to the cancer-specific phenotypic changes.Citation1,2
Increased expression of one or more oncogenic regulators is consistently observed in different cancers. Overexpressed oncogenic factors, in most cases, directly contribute to the carcinogenesis process by activating various genes involved in cell cycle-regulation, cell motility, adhesion and signaling pathways. Gene specificity is usually achieved via selective recruitment to gene regulatory regions, which is elicited through binding to specific DNA sequences or interactions with other promoter-bound factors.
In some cases, however, the recruitment of oncogenic regulators is not limited to a specific set of genes. For example, recent studies suggested that the Myc oncogene is universally recruited to the regulatory regions of all active genes.Citation3,4 As Myc protein levels rise, it is loaded quantitatively onto active promoters and enhances transcription. These findings suggested that Myc acts as an amplifier of existing active transcription rather than as a factor driving a dedicated cancer-specific gene expression program. Amplification of a pre-existing cancer-specific gene expression pattern may contribute to aggressive, uncontrolled growth. It can also provide an advantage for the adaptation to the negative effects of defective cellular pathways, invariably observed in most cancer cells.Citation3,4
Up until recently, Myc was the only known oncogene acting universally at all active genes. This feature however is not unique for Myc. We recently described a similarly promiscuous genome-wide recruitment to active regulatory regions for Smyd3, in a mouse model of hepatocellular carcinoma (HCC).Citation5 Smyd3 is a histone methyltransferase, highly upregulated in several tumors, including lung and pancreatic adenocarcinomas,Citation6 colorectal cancer and HCC.Citation5 In human HCC patients, Smyd3 expression has diagnostic and prognostic value. It is among the highest-ranking genes whose overexpression can discriminate cancerous from normal livers and its mRNA levels negatively correlate with the overall survival of the patients and the probability of tumor grade progression or new tumor occurrence after chemo-surgery.Citation5 Investigating mouse models developing liver or colon cancer in response to carcinogenic agents, we found that Smyd3 expression is required for tumor formation. Gene expression analyses in experimentally induced liver cancer revealed that Smyd3 function is essential for the generation of HCC-specific gene expression program. Smyd3 enhances cell proliferation capacity via the activation of cell cycle regulators, other oncogenes like Myc, Ctnnb1, components of Jak-Stat3 pathway and several genes involved in epithelial-mesenchymal transition (EMT). Acting on multiple pathways and targets at different levels of regulatory hierarchy, Smyd3 function creates a self-perpetuating circuitry leading to intensified proliferation effects.Citation5
Mechanistic insights into the Smyd3-dependent regulation of cancer-specific gene expression patterns came from genome-wide analyses of Smyd3 recruitment to its target genes. Surprisingly, Smyd3 was detected in more than 10 thousand promoter regions in HCC, the majority of which was co-occupied by RNA Polymerase-II and H3K4Me3-containing nucleosomes. This is highly reminiscent to the recruitment pattern of Myc described above. The highly promiscuous invasion of active promoters is probably mediated by the high affinity association of Smyd3 with H3K4Me3-containing histone tails and its ability to interact directly with RNA Polymerase-II. Smyd3 binding density at the core promoter regions positively correlated with the density of RNA Polymerase-II and with overall transcriptional outputs, suggesting that, similar to Myc, Smyd3 acts as an “amplifier” or “potentiator” of transcription of the already active genes.
Nevertheless, there is a fundamental difference between Smyd3 and Myc actions as revealed by the functional outcomes of their recruitment ( scheme). Unlike the universal effect of Myc, the expression of only a small fraction (590 out of 10300) of the Smyd3-occupied genes was induced in a Smyd3-dependent manner.Citation5 Insight into the differential regulatory potential of promoter-bound Smyd3 is provided by Kernel density estimation analysis of Smyd3 binding as assessed by ChIP-seq tag densities. Smyd3 binding displayed a bimodal Kernel density pattern, characteristic of transcription factors with high- and low-affinity locations.Citation5 In contrast, the histogram of Myc density was unimodal, compatible with continuous, “analog” target selection mechanism.Citation3 This prompted us to further analyze the distribution of Smyd3-regulated genes and compare them with the tag density profile of all Smyd3-bound locations. As shown in graph, Smyd3-regulated genes are mainly distributed to tag density points under the smaller peak at the right side of the bimodal curve, corresponding to high affinity sites. Likewise, the majority of the genes falling under the peak corresponding to “low affinity” sites were not responsive to Smyd3.
Figure 1. Schematic presentation of the transcriptional effects on target promoters. Both Smyd3 and Myc invade already active gene regulatory regions. Smyd3 amplifies the transcription of specific genes only, while Myc mediates transcription amplification of all invaded genes. Graph shows Gaussian Kernel density estimation based on the distribution of Smyd3 reads per kb per million reads (RPKM) in promoter regions (-10 kb to + 1kb from the TSS) in natural logarithm (ln) scale in all Smyd3-bound genes (green line) and in genes bound by Smyd3 and up-regulated in HCC in a Smyd3-dependent manner (red line). The bimodality of the Kernel density estimation in the promoters of all Smyd3-bound genes is indicative of Smyd3 bindings with higher (mode at right) and lower (mode at left) affinities. Smyd3-regulated genes are associated with higher affinity Smyd3 binding sites.
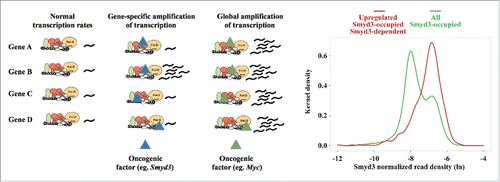
These results suggest that the binding stability and/or residence time of Smyd3 at a given promoter region can greatly influence its regulatory function. We, therefore, propose that promoter context plays a determinative role in Smyd3-mediated gene regulation. The highly restricted functional specificity of Smyd3 most likely comes from its ability to synergize with a specific set of transcription factors, which activate cancer-related genes.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Sarris M, et al. Oncogene 2014; 33:1207-17; PMID:23503463; http://dx.doi.org/10.1038/onc.2013.87
- Nikolaou K.C. et al. EMBO J. 2015; 34:430-47; PMID:25515659; http://dx.doi.org/10.15252/embj.201489279
- Nie Z. et al. Cell 2012; 151:68-79; PMID:23021216; http://dx.doi.org/10.1016/j.cell.2012.08.033
- Lin CY. et al. Cell 2012; 151:56-67; PMID:23021215; http://dx.doi.org/10.1016/j.cell.2012.08.026
- Sarris EM. et al. Cancer Cell 2016; 29:354-66; PMID:26908355; http://dx.doi.org/10.1016/j.ccell.2016.01.013
- Mazur PK. et al. Nature 2014; 510:283-7; PMID:24847881; http://dx.doi.org/10.1038/nature13320