
ABSTRACT
In eukaryotes, bulk histone expression occurs in the S phase of the cell cycle. This highly conserved system is crucial for genomic stability and proper gene expression. In metazoans, Stem-loop binding protein (SLBP), which binds to 3′ ends of canonical histone mRNAs, is a key factor in histone biosynthesis. SLBP is mainly expressed in S phase and this is a major mechanism to limit bulk histone production to the S phase. At the end of S phase, SLBP is rapidly degraded by proteasome, depending on two phosphorylations on Thr 60 and Thr 61. Previously, we showed that SLBP fragment (aa 51–108) fused to GST, is sufficient to mimic the late S phase (S/G2) degradation of SLBP. Here, using this fusion protein as bait, we performed pull-down experiments and found that DCAF11, which is a substrate receptor of CRL4 complexes, binds to the phosphorylated SLBP fragment. We further confirmed the interaction of full-length SLBP with DCAF11 and Cul4A by co-immunoprecipitation experiments. We also showed that DCAF11 cannot bind to the Thr61/Ala mutant SLBP, which is not degraded at the end of S phase. Using ectopic expression and siRNA experiments, we demonstrated that SLBP expression is inversely correlated with DCAF11 levels, consistent with the model that DCAF11 mediates SLBP degradation. Finally, we found that ectopic expression of the S/G2 stable mutant SLBP (Thr61/Ala) is significantly more toxic to the cells, in comparison to wild type SLBP. Overall, we concluded that CRL4-DCAF11 mediates the degradation of SLBP at the end of S phase and this degradation is essential for the viability of cells.
Introduction
Eukaryotic S phase is characterized by chromosome replication, which requires synthesis of DNA and sufficient amount of histone proteins. Cells need to achieve very delicate balance between DNA synthesis and histone levels, and confine the bulk histone production to the S phase where the DNA amount is doubled. Imbalance between DNA and histone synthesis may cause improper gene expression, genomic instability and cell death.Citation1-3
In metazoans, canonical histones are encoded by so-called replication dependent histone genes. These multi-copy genes lack introns and their mRNAs end in a conserved stem-loop structure instead of a polyA tail. Other than the 5′ capping, the only processing reaction required to produce mature histone mRNAs is an endolytic cleavage after the conserved stem-loop structure at their 3′ end.Citation4 Histone mRNA expression is limited to the S phase and this is regulated by transcriptional and posttranscriptional mechanisms. At the G1-S transition, the transcription of histone genes increases along with a significant increase in the pre-mRNA processing efficiency, due to the increase in SLBP.Citation5-8 Together these account for the rapid increase in the level of histone mRNAs as cells enter the S phase. At the end of S phase, both the histone pre-mRNA processing and half-life of histone mRNAs are diminished in order to rapidly shut down histone mRNA expression.Citation4,7
Stem-loop binding protein (SLBP) binds to the conserved stem-loop structure at the 3′ ends of histone mRNAs and mediates all aspects of histone mRNA metabolism including pre-mRNA processing, nuclear export, translation and stability.Citation9,10 SLBP is present at high levels only in S phase, and this is a major mechanism to limit canonical histone production to the S phase.Citation4,8 SLBP expression is cell cycle regulated without a significant change in its mRNA level.Citation11 In G1, SLBP is kept low by coordinate action of translation control and proteasome mediated degradation.Citation12 At late G1, SLBP expression significantly increases and reaches to the highest level in S phase. At the end of S phase (S/G2), SLBP is rapidly degraded by the proteasome to shut down histone production.Citation8,11 Degradation of SLBP at the end of S phase requires specific phosphorylations of two threonines in N-terminal TTP (aa 60–62) motif.Citation8 The initial trigger for the late S phase degradation of SLBP is Thr 61 phosphorylation by cyclin A/Cdk1,Citation13 which emerges to be important regulator of S-G2 transition.Citation14-17 The phosphorylation of Thr 61 depends on a downstream cyclin binding motif which is critical for the proper recruitment of cyclin A/Cdk1 to SLBP. Once Thr 61 is phosphorylated, it primes the subsequent phosphorylation of Thr 60 by CK2 and the doubly phosphorylated SLBP is recognized by an unknown E3 ligase for the proteasome mediated degradation.Citation13,18
The ubiquitin-proteasome system is the major pathway by which cells target proteins for specific and time-controlled degradation. Ubiquitin, a small protein highly conserved from yeast to mammals, is covalently conjugated to proteins via a cascade of 3 enzymatic activities. Ubiquitin is initially bound and activated by an E1 ubiquitin activating enzyme in an ATP-dependent manner and subsequently, it is transferred to an E2 ubiquitin conjugating enzyme. Finally, an E3 ligase recognizes a specific substrate protein and bridge the substrate and E2 to promote ubiquitin ligation. Once a polyubiquitin chain is formed on the target protein, it is recognized by 26S proteasome for degradation. Cullin-RING-based E3s (CRLs) constitute a major subclass of E3 ligase complexes. In humans, 7 different Cullins (Cul 1, 2, 3, 4A, 4B, 5, 7) have been identified, and each functions as a “scaffold” to assemble a CRL complex.Citation19 Mammalian cells express two closely related Cul4 paralogues Cul4A and Cul4B. Cul4s bind to DDB1 protein through their N-termini. DDB1 functions as an adaptor protein to recruit a DCAF (DDB1-Cul4 associated factors) protein, which functions as the substrate receptor of the complex to specifically recognize and bind to the target proteins.Citation20,21 Cul4-based E3 ligases (CRL4) are implicated in the regulation of chromatin biology including DNA damage response, histone modification, and nucleosome assembly.Citation22-24 Depletion of Cul4A results in chromatin dysfunctions in yeast and mammalian cells, and notably over expression of Cul4A has been reported in many cancer types.Citation25 There are several studies reporting the critical roles of CRL4s in the cell cycle, especially in S phase related degradations. CRL4s have been shown to target several key players including Cdt1, p21, p27, E2f1, Set8 and Chk1 for degradation, in order to maintain proper S phase and S-G2 progression Citation23,25-27.
Previously, we showed that SLBP fragment (between amino acids 51–108) with just Thr 60 and Thr 61 as possible phosphoacceptor sites is sufficient to mediate the degradation of SLBP at the end of S phase. When this SLBP fragment was fused to GST, just like endogenous full-length SLBP, it was degraded at the end of S phase depending on Thr 60 and Thr61 phosphorylations.Citation13 Here, we used this phosphorylated fusion protein as a bait, to pull-down the unknown E3 ligase that mediates the S/G2 degradation of SLBP. Based on our results, we proposed that Cul4A-DCAF11 E3 ligase mediates the degradation of SLBP at the end of S phase, to inhibit histone synthesis.
Results
GST-SLBP fragment fusion protein is sufficient to mimic the S/G2 degradation of SLBP depending on two phosphorylations on Thr 60 and Thr 61.Citation13 Here, we produced the phosphorylated form of this fusion protein, in order to pull-down the unknown E3 ligase that mediates the S/G2 degradation of SLBP. We bacterially expressed and purified the GST-SLBP fragment fusion protein, and as in our previous study, we in vitro phosphorylated Thr 61 and Thr 60 by cyclin/Cdk1 and CK2, respectively. We also prepared Thr 61 and Thr 60 changed to alanine mutant version, which is known to be stable at the end of S phase.Citation13 Using these fusion proteins, and GST, as baits, we performed pull-downs from the lysate of HeLa cells synchronized by double-thymidine method and collected at the end of S phase. We ran the results on SDS-PAGE, and silver stained (, Fig. S1). We concentrated on the bands that came down with phosphorylated GST-SLBP fragment, but not with S/G2 stable mutant or GST alone, and looked for a protein with potential E3 ligase role. We tested several different buffers, wash and staining conditions. With the pull-down conditions explained in the materials and methods, and increased gel staining, we repeatedly detected a faint band (∼65 kDa), present only in the pull-downs by phosphorylated SLBP fragment, but not in the others (Fig. S1C). We sent the band to mass spectrometry analysis (LC-MS/MS), and repeatedly got DCAF11 with high mascot score (). DCAF11 protein was shown to bind CRL4 complex and was proposed as a substrate recognizing subunit of the complex.Citation20,21 Based on these results, we decided to pursue DCAF11 as a candidate protein that mediates SLBP degradation at the end of S phase.
Figure 1. Experimental setup for pull-downs. (A) Amino acid sequence of SLBP fragment, sufficient to mediate S/G2 degration of SLBP. All the possible phosphoacceptor sites other than Thr 60 and Thr 61 were changed into alanine and were shown in light color. Mutations for the S/G2 stable version were shown below. (B) Schematic illustration of baits and experimental setup. (C) Amino acid sequence of DCAF11 with the peptides recovered in LC-MS/MS.
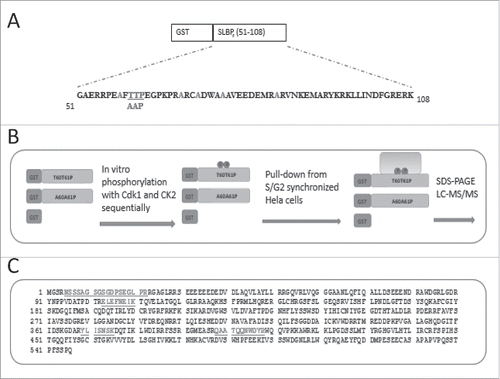
Next, we validated our results with pull-downs followed by western blot analysis. We ectopically expressed HA-tagged DCAF11 and by western blot we detected HA-DCAF11 expression at expected size. Using lysates from those cells, we performed pull-downs followed by western blot analysis. In line with our mass spectrometry data we repeatedly showed that HA-DCAF11 came down with phosphorylated GST-SLBP fragment (). We detected none or very little amount of HA-DCAF11 in the pull-downs with either unphosphorylated or S/G2 stable mutant (Thrs to Ala) versions of the bait (). Our results confirm that DCAF11 interacts with SLBP fragment depending on the phosphorylations that are required for the degradation of SLBP at the end of S phase.
Figure 2. DCAF11 and Cul4A were pulled down by GST-SLBP fragment depending on phosphorylations on Thr 60 and Thr 61. HeLa cells were transfected with HA-DCAF11 (A) or Myc-Cul4A (B) and collected 48 hrs after transfection. Cells were lysed and pull-downs were performed with the indicated baits as explained in the materials and methods. GST-SLBPF (pTpTP) indicates phosphorylations on Thr 60 and Thr 61. Pull-downs were run on SDS-PAGE and immunoblotted for HA-DCAF11 or Myc-Cul4A. The amounts of baits were assessed by Comassie Briliant Blue (CBB) staining.
In the CRL4 complex, DCAF11 was proposed to be the substrate receptor that recruits the target proteins to Cul4. We also checked whether our bait could pull-down Cul4A. We transiently expressed Myc-Cul4A and repeated similar pull-down experiments with our baits. With western blot analysis, we detected significant amount of Myc-Cul4A that came down with phosphorylated GST-SLBP fragment but not with unphosphorylated, S/G2 stable mutant version (). Our results show that SLBP fragment interacts with both DCAF11 and Cul4A depending on phosphorylations, which are known to be required for SLBP degradation at the end of S phase.
Ectopic expression of HA-DCAF11 induces proteasome mediated degradation of SLBP
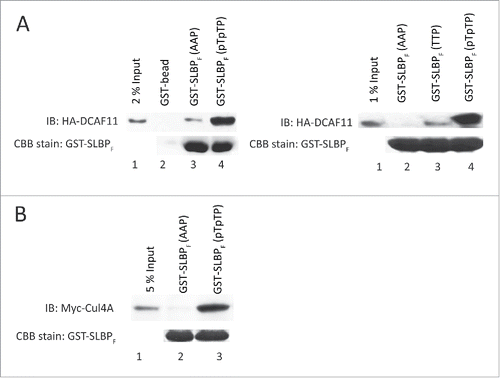
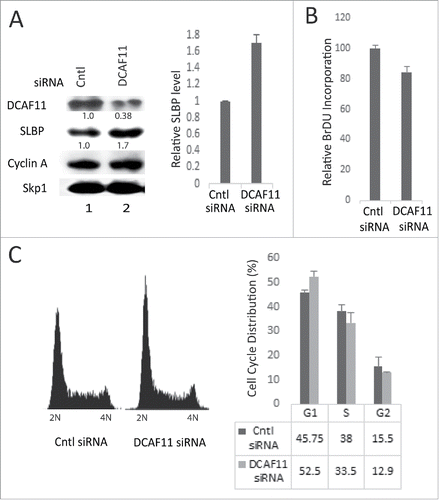
Next, we checked the levels of SLBP in response to ectopic expression of HA-DCAF11 and detected a significant decrease in the SLBP expression (). Further, when we treated the cells with proteasome inhibitor (MG132) in the last 2 hours before the collection, SLBP levels were increased, suggesting that ectopic expression of DCAF11 induces proteasome mediated degradation of SLBP ().
Figure 3. Ectopic expression of DCAF11 induces proteasome mediated degradation of SLBP. (A) HeLa cells were transfected with the empty vector (EV) or HA-DCAF11 construct and collected 48 hrs after transfection. Cells were lysed and whole cell extracts were immunblotted for HA-DCAF11, SLBP and Skp1 (as a loading control). SLBP levels were quantified and the level in the EV cells was set to 1. In the right panel, results from 3 independent experiments were graphed as Mean ± SD. (B) HeLa cells were transfected with EV or HA-DCAF11 construct. 48 hours after transfection, BrdU was introduced into the cultures for 2 hours and BrdU incorporation levels were quantified using colorimetric detection kit as explained in materials and methods. Mean BrdU incorporation values (n = 3) ± SD were graphed as a percentage of the levels detected in the EV transfected cells. (C) Cell cycle profiles of the cells were determined by PI (Propidium Iodide) staining followed by Flow Cytometry analysis. Right pannel shows the mean ± SD of 3 independent experiments. (D) Cells were transfected with EV or HA-DCAF11 and collected 48 after the transfection. In the lane 3, protesome inhibitor (MG132) was added for the last 2 hrs before collection. Cells were lysed and immunblotted for HA-DCAF11, SLBP and Skp1.
We also determined the effect of DCAF11 ectopic expression on DNA replication and the cell cycle. 46 hours after the transfection of either empty vector or HA-DCAF11 construct, we labeled the cells with BrdU for 2 hours and determined the total BrdU incorporation levels using colorimetric BrdU detection kit. In each culture, we also quantified the viable cell amount using Wst-1 assay and normalized the BrdU incorporation levels to the viable cell numbers, in order to exclude the possible interfering effects of cell number differences. We detected a significant increase in the BrdU incorporation in the HA-DCAF11 transfected cells (). In order to determine the cell cycle distribution, we also performed propidium iodide staining followed by flow cytometry analysis. In response to ectopic expression of HA-DCAF11, we detected a significant accumulation of cells in the early S phase (). When considered together, our results suggest that HA-DCAF11 expressed cells entered S phase and initiated DNA replication, however, most of them accumulated in the early S phase due to a significant delay in early S phase progression (), as is the case in previous SLBP knockdown studies.Citation28
SLBP interacts with both DCAF11 and Cul4A
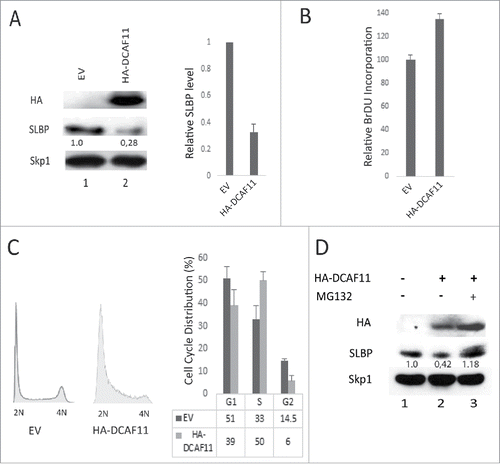
Next, we determined whether DCAF11 interacts with full-length SLBP with co-immunoprecipitation experiments. We ectopically expressed HA-DCAF11 along with hisSLBP. In order to prevent the DCAF11 induced decrease in SLBP expression, we treated the cells with proteasome inhibitor in the last 2 hours before the collection. We performed immunoprecipitation with SLBP antibody and found that HA-DCAF11 is pulled down along with SLBP (). We also performed the reverse co-immunoprecipitation experiment, where we immunoprecipitated HA-DCAF11 with HA antibody and detected both his-tagged and endogenous SLBP in the immunoprecipitate, confirming the SLBP and DCAF11 interaction ().
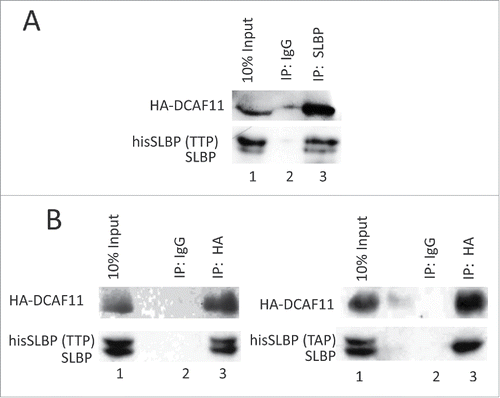
Figure 4. DCAF11 interacts with wild type, but not Thr 61/Ala mutant SLBP. (A) HeLa cells were transfected with HA-DCAF11 along with hisSLBP. Cells were treated with MG132 for the last 4 hrs before collection. Cells were lysed and immunoprecipitations with nonspecific IgG or anti-SLBP were performed. Whole cell extract (input) and immunoprecipitates were analyzed by western blot for indicated proteins. (B) HeLa cells were transfected with HA-DCAF11 along with wild type hisSLBP (TTP) or Thr61/Ala mutant hisSLBP (TAP). Whole cell extracts (input) were subjected to immunoprecipitation (IP) with either nonspesific IgG or anti-HA and immunoprecipitates were immunoblotted for indicated proteins.
We also determined whether Cul4A and SLBP can be detected in the same complex. HeLa cells were co-transfected with Myc-tagged Cul4A, HA-DCAF11 and hisSLBP and immunoprecipitation was performed with SLBP antibody. When we immunoprecipitated SLBP, we detected Myc-Cul4A in the immunoprecipitate (), showing that SLBP and Cul4A were in the same complex. In the immunoprecipitates with anti-SLBP, we detected a slower migrating band, which is probably the neddylated version of Cul4A present in active CRL4 complexes. We also did the reverse co-immunoprecipitation experiment, where we immunoprecipitated Myc-Cul4A and found both his-tagged and endogenous SLBP in the immunoprecipitate (). Our results showed that SLBP and Cul4A are present in the same complex, consistent with the potential role of CRL4 in the regulation of SLBP expression ().
Figure 5. SLBP interacts with Cul4A. HeLa cells were transfected with Myc-Cul4A, HA-DCAF11 and hisSLBP. Cells were treated with MG132 for the last 4 hours before collection. Cells were lysed and immunoprecipitations with either nonspecific IgG, anti-SLBP (A) or anti-Myc (B) were performed. Whole cell extracts (input) and immunoprecipitates were analyzed with western blot for indicated proteins.
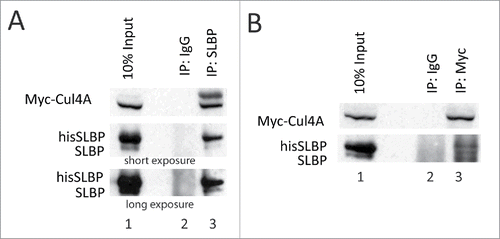
DCAF11 interacts with wild type, but not Thr 61/Ala mutant SLBP
Phosphorylation of Thr 61 by cyclin A/Cdk1 is known to prime the subsequent phosphorylation of Thr 60 by CK2 and doubly phosphorylated SLBP is recognized by an unknown E3 ligase for the degradation at the end of S phase.Citation13,18 Mutation of Thr 61 into Ala prevents this degradation.Citation8 The DCAF11 was found in the pull-downs by the SLBP fragment doubly phosphorylated on Thr 60 and Thr 61. Based on these, we checked whether Thr 61 is required for full-length SLBP and DCAF11 interaction. We co-expressed either wild type or S/G2 stable mutant (Thr 61 to Ala) hisSLBP together with HA-DCAF11 and immunoprecipitated with HA antibody. In line with the GST pull-down assays, we showed that endogenous and ectopically expressed wild type SLBP, but not the Thr 61/Ala mutant version, came down with HA-DCAF11 (), suggesting that DCAF11 recognizes SLBP depending on Thr 61 phosphorylation, which is known to trigger S/G2 degradation of SLBP.
RNAi based knockdown of DCAF11 leads to an increase in SLBP levels and impairs Cul4A binding
In order to further examine the role of DCAF11 in regulation of SLBP expression, we knocked down DCAF11 in HeLa cells using DCAF11-specific siRNA and checked the SLBP level with western blot analysis. In our knockdown experiments, we obtained a decrease in DCAF11 levels by roughly 60%. However, we reproducibly saw a significant increase in SLBP levels (). In the DCAF11 siRNA transfected cells, we also observed a decrease in BrdU incorporation (). When we performed propidium iodide staining followed by flow cytometry analysis, we detected a small, but significant increase in G1 and a decrease in S phase cells. Our results imply that, besides its role in SLBP degradation, DCAF11 is critical for S phase entry and DNA replication ().
Figure 6. Knockdown of DCAF11 induces SLBP expression. (A) HeLa cells were transfected with control (non-targeting) or DCAF11 specific siRNA, and collected 48 hrs after transfection. Cells were lysed and whole cell extracts were immunoblotted for DCAF11, SLBP, Cyclin A and Skp1. SLBP levels were quantified and the level in the control cells was set to 1. Results from 3 independent experiments were graphed as mean ± SD in the panel on the right. (B) BrdU incorporation levels were quantified as explained in the materials and methods using colorimetric detection kit. Mean values (n = 3) ± SD were graphed as a percentage of the control siRNA transfected cells (C) Cell cycle profiles of the cells were determined by PI staining followed by Flow Cytometry analysis. In the right panel, results from 3 independent experiments were graphed as mean ± SD.
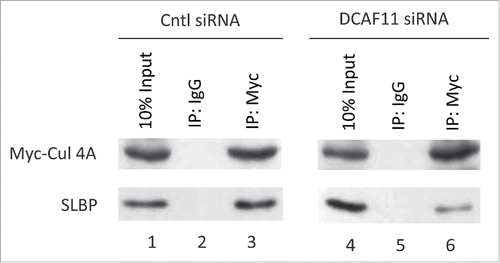
We also checked whether knockdown of DCAF11 impairs Cul4A and SLBP interaction. We transfected cells with Myc-Cul4A construct along with control or DCAF11-specific siRNA. Next, we immunoprecipated the Myc-Cul4A and checked whether SLBP came down with Myc-Cul4A. Although we immunoprecipitated comparable amount of Myc-Cul4A, we detected significantly less SLBP in the immunoprecipitates from the DCAF11 siRNA transfected cells (). Our results corroborate the model that DCAF11 binds and recruits SLBP to Cul4A.
Figure 7. Cul4A and SLBP interaction is impaired by DCAF11 RNAi. HeLa cells were transfected with Myc-Cul4A along with control (non-targeting) or DCAF11 spesific siRNAs. Cells were lysed and immunoprecipitations with non-spesific IgG or anti-Myc were performed. Whole cell extracts (input) and immunoprecipitates were analyzed by western blot for indicated proteins.
Thr61/Ala mutant SLBP induces cell death in HeLa cells
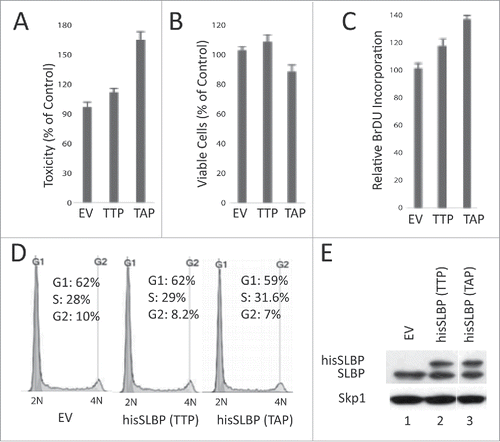
The TTP motif (containing Thr 60 and Thr 61), which regulates the S/G2 degradation of SLBP, is highly conserved in vertebrates, suggesting the importance of this regulation. In order to determine whether S/G2 degradation of SLBP is critical for the cells, we transiently expressed wild type or S/G2 stable mutant hisSLBP (Thr 61 to Ala) in similar levels (), and compared their possible effects on the viable cell number, the cell death and DNA replication (). We assessed the viable cell number using Wst-1 cell viability assay, and determined cell death levels using LDH cytotoxicity assay, which quantifies the level of cell death by measuring the released Lactate dehydrogenase (). We repeatedly found that S/G2 mutant version was significantly more toxic to the cells, however, the actual dead cell amount induced in our experiments appeared to be limited (). In line with that, we detected a modest decrease in the viable cell number in S/G2 stable mutant SLBP expressed cells (). We also determined the effects on DNA replication and cell cycle distribution. We found that ectopic expression of SLBP induces an increase in the BrdU incorporation rate (). When we performed the propidium iodide staining followed by flow cytometry analysis, we didn't detect an obvious effect on the cell cycle distribution of the cells (). Overall, we observed a limited difference between the cell cycle related effects of wild-type and S/G2 stable mutant SLBP, however, our toxicity results suggest that S/G2 degradation of SLBP is important for the viability of cells.
Figure 8. S/G2 stable mutant SLBP induces cell death in HeLa cells. HeLa cells were transfected with EV, wild type hisSLBP (TTP) or S/G2 stable mutant (Thr 61/Ala) hisSLBP (TAP). (A) 48 hrs after the transfection, cell death levels were assessed by LDH release assay, (B) cell viability was quantified using Wst-1 viability assay, and (C) BrdU incorporation levels were quantified as explained in the materials and methods using colorimetric detection kit. Mean (n = 3) ± SD were graphed as a percentage of the values detected in the EV transfected cells. (D) Cell cycle profiles of the cells were determined by PI staining, followed by Flow Cytometry analysis. (E) Cells were lysed and whole cell extracts were immunoblotted for SLBP and Skp1.
Discussion
CRL4-DCAF11 mediates the late S phase degradation of SLBP
SLBP is mainly expressed in S phase and this is a major mechanism to limit histone production to the S phase.Citation4,8 At the end of S phase, SLBP undergoes proteasome mediated degradation triggered by sequential phosphorylations of Thr 61 and Thr 60 by cyclin A/Cdk1 and CK2, respectively.Citation13,18 Doubly phosphorylated SLBP is recognized by an unknown E3 ligase for the late S phase degradation. Previously, we showed that when fused to GST, the N-terminal SLBP fragment (aa 51-108) with only Thr 60 and 61 as potential phosphoacceptor sites, is sufficient to mimic S/G2 degradation of SLBP in HeLa cells. Further, as in full-length SLBP, when we mutated either of the threonines to alanine, GST-SLBP fragment fusion protein became stable at the end of S phase.Citation13 Here, we used this GST fusion protein, containing the SLBP fragment (aa 51-108) that is sufficient to mediate S/G2 degradation, to pull-down unknown E3 ligase responsible for the degradation of SLBP at the end of S phase. We bacterially expressed and purified this GST-SLBP fragment fusion protein and in vitro phosphorylated on Thr 60 and Thr 61. We also produced the S/G2 stable mutant version, where those threonines were converted to alanine in order to prevent the phosphorylations required for S/G2 degradation. Using these proteins as baits, we performed pull-down experiments from late S phase HeLa cells' lysates, followed by mass spectrometry analysis (). According to our model Thr 60 and Thr 61 phosphorylations on SLBP are the regulators of the SLBP degradation at the end of S phase. However, it was also possible that there might be a cell cycle dependent regulation on the unknown E3 ligase that we were searching for. Therefore, in our initial pull-downs, we used lysates of the cells collected at the end of S phase, where endogenous SLBP is degraded and the E3 ligase should be active. In the pull-downs by the phosphorylated GST-SLBP fragment, we repeatedly found DCAF11 by mass spectrometry analysis. Next, we also validated our results by western blot analysis. We performed pull-downs from HA-DCAF11 transfected cells and showed that HA-DCAF11 was pulled down by phosphorylated GST-SLBP fragment, but not with unphosphorylated or S/G2 stable mutant versions (). Previously, DCAF11 was shown to bind CRL4 and was proposed as a substrate recognizing subunit of CRL4 complex.Citation20,21 In line with that, we showed that Cul4A is also pulled down by GST-SLBP fragment again depending on Thr 60 and Thr 61 phosphorylations (). Since we found DCAF11 and Cul4A in the pull-downs by the phosphorylated SLBP fragment that mediates the S/G2 degradation of SLBP, we decided to follow CRL4-DCAF11 as the candidate E3 complex, responsible for the SLBP degradation at the end of S phase.
Next, with co-immunoprecipitation experiments, we confirmed the interaction of DCAF11 and full-length SLBP in the lysate (). Parallel with our pull-down experiments, we repeatedly showed that DCAF11 binds to wild type but not to S/G2 stable mutant (Thr 61 to Ala converted) SLBP, confirming that Thr 61 is required for DCAF11 and SLBP interaction (). Since the phosphorylation of Thr 61 is required for S/G2 degradation of SLBP, disruption of DCAF11 and SLBP interaction by Thr 61 to Ala mutation fits with the model that DCAF11 is the mediator of S/G2 degradation of SLBP.
We also determined whether we can detect Cul4A and SLBP in the same complex. When we performed co-immunoprecipitation experiments from proteasome inhibitor treated HeLa cells, we detected Cul4A and SLBP in the same complex, consistent with our model that DCAF11 recruits SLBP to CRL4 to mediate its degradation ().
Cul4A itself is regulated, and in the active E3 ligase complex the neddylated form (NEDD8 conjugated) is present.Citation29 Consistent with that, in the immunoprecipitate with the SLBP antibody, we detected a slower migrating Myc-Cul4A band, which appeared as a faint band in the input (). This enriched band is probably the neddylated form of Cul4A, which is required for the fully active Cul4A-based E3 ligase complex.
We also determined whether knockdown of DCAF11 will impair the Cul4A and SLBP interaction. We showed that in the DCAF11 siRNA transfected cells, co-immunoprecipitation of SLBP with Cul4A were significantly inhibited (). Our result further strengthens the model that DCAF11 recruits SLBP to Cul4A-based complex.
In line with our model, we also demonstrated that SLBP expression is inversely correlated with DCAF11 levels by ectopic expression and siRNA experiments. We showed that the ectopic expression of HA-DCAF11 triggered proteasome mediated degradation of SLBP (). Further, when we knocked down DCAF11, again fitting with our model, we detected a significant increase in the SLBP expression levels ().
We determined the effect of DCAF11 ectopic expression on the BrdU incorporation, and detected an increase in the BrdU incorporation levels (). We also determined the cell cycle distribution of those cells, and observed a significant accumulation of the cells in early S phase suggesting an early S phase delay (). It appears that although the DNA replication rate of individual cells was impaired causing an early S phase delay and accumulation, since most of the cells in the culture were in the S phase and started some level of DNA replication (thus BrdU incorporation), we detected an increase in the BrdU incorporation ().
When we knocked down DCAF11, we detected a decrease in the BrdU incorporation. We also detected an increase in the G1 cells (). It appears that in our knockdown experiments, the remaining level of DCAF11 was sufficient to prevent a stronger BrdU incorporation decrease and G1/S arrest. However, our results suggest that DCAF11 has critical functions in S phase entry and DNA replication, which are presumably distinct than its role in SLBP degradation at the end of S phase.
Taken together, DCAF11 appears to be essential for S phase entry and progression. However, after the ectopic expression of DCAF11, we detected an early S phase delay similar to previously reported SLBP knockdown studies. It appears that while DCAF11 overexpression pushes the cells to the S phase, the significant decrease in the SLBP level causes an early S- phase delay, probably because of slowed rate of histone synthesis, just like the effect seen in SLBP knockdown studies.Citation28,30
SLBP is expressed in S phase. However, when we expressed HA-DCAF11, despite the increase in S phase cells, SLBP levels significantly decreased. Similarly, although DCAF11 siRNA had inhibitory effect on S phase entry, SLBP levels went up significantly in those cells. In both experiments the SLBP levels don't change in line with the changes in the percentage of S phase cells. These findings suggest that the effects on SLBP expression in overexpression and knockdown experiments were not due to the indirect effects of the cell cycle profile changes, but possibly due to the direct regulation of SLBP by DCAF11.
S/G2 stable mutant SLBP is toxic to the cells
The motifs that control S/G2 degradation of SLBP (TTP and downstream cyclin binding site) are conserved in vertebrates. Moreover, when we expressed either XSLBP1 or mSLBP in HeLa cells, both are also degraded at the end of S phase (Koseoglu, MM and Marzluff, WF, unpublished data). The possible conservation of S/G2 degradation mechanism suggests that it is crucial for the cells. In order to determine the possible effects of inhibiting the S/G2 degradation of SLBP, we transiently expressed wild type and S/G2 mutant (Thr 61/Ala) SLBP at similar protein levels and analyzed their effects on BrdU incorporation, living cell numbers and cell death (). Our BrdU incorporation results suggest that DNA replication rate increases in response to ectopic expression of SLBP, which is probably due to the previously identified role of new histone supply on regulation of replication fork speed.Citation31 Proper regulation of histone synthesis is also critical for the cell viability.Citation1 When we determined the cell death level, we reproducibly observed a significantly higher toxic effect of S/G2 stable mutant SLBP, than the wild-type (). We also quantified the living cell numbers, which reflects both cell division and cell death rate at the same time, and detected a modest decrease in the viable cell number in response to S/G2 stable SLBP expression (). It appears that the actual amount of cell death, we detected in HeLa cells is limited. However, the reproducible induction of significantly higher cell death by S/G2 stable mutant SLBP, than the wild-type, implies that SLBP degradation at the end of S phase is critical for the cell viability. Overall, our result suggests that preventing the S/G2 degradation of SLBP induces cell death, and the mechanisms and possible involvement of checkpoints are yet to be investigated.
CRL4s as emerging power of S phase
In recent years, several studies revealed significant roles for Cul4-based E3 ligase complexes in the cell cycle progression, especially by mediating the S phase related degradations of several key regulatory proteins. CRL4Cdt2, which emerged as critical regulator of cell cycle, was found to mediate S phase degradations of several key players including Cdt1, p21, Set8, Chk1.Citation25 CRL4Cdt2 was also linked to proper expression of histone genes by mediating the degradation of Set8. CRL4Cdt2 mediated S phase degradation of Set8 was shown to be required for transcription of histone genes.Citation32 DCAF11 was previously shown to bind CRL4 and was proposed as a substrate recognizing subunit of the CRL4 complex.Citation20 However, there was almost no information about its targets. The only related study was in C. elegans, where the ortholog of DCAF11 was proposed to be involved in the regulation of the transcription factor SKN-1, which protects animals from stress and promotes longevity.Citation33 In our study, we concluded that CRL4DCAFCitation11 mediates the degradation of SLBP at the end of S phase, which is an important mechanism to coordinate cell cycle and histone biogenesis, and proposed a role for CRL4DCAFCitation11 in the mammalian system.
Conclusion
SLBP is the major factor that regulates all aspects of histone mRNA metabolism. Restriction of SLBP expression to the S phase is a major mechanism to limit canonical histone production to the S phase. SLBP is rapidly degraded at the end of S phase as a mechanism to shut down canonical histone production. Here, we proposed that CRL4DCAFCitation11 mediates the degradation of SLBP at the end of S phase and this degradation is critical for the cell viability. Overall, our study revealed a significant role for CRL4DCAF11 in regulation of histone biosynthesis and introduced a new component to the emerging S phase related roles of CRL4s.
Materials and methods
GST-SLBP fragment fusion protein
The GST-SLBP fragment construct for bacterial expression was prepared as explained in Koseoglu et al., 2008.Citation13 Roughly, the SLBP fragment (aa 51-108) was sub-cloned into the pGEX2T vector just after the glutathione S-transferase (GST) tag using a PCR-generated insert with Eco-RI and Bam-HI sites. All phosphoacceptor amino acids except Thr60 and Thr61 were converted into alanine using Quickchange site directed mutagenesis protocol using appropriate primers (Stratagene). For bacterial expression, the constructs were transformed into BL21 (DE3) cells. The expression of GST-SLBP fragment in BL21 (DE3) cells was induced by 0,75 mM IPTG for 2 hours at 37°C. Induced cells were lysed by sonicating on ice for 4 times. Lysates were clarified by centrifugation for 10 min at 14,000 rpm at 4°C. GSH beads were added to the lysate and rocked at 4°C for 2 hrs. Purified proteins were analyzed by SDS-PAGE and silver staining. For pull-down experiments, GST-SLBP bound to the beads was used.
Phosphorylation of the baits
Sequential phosphorylations of the threonines were performed using bacolovirus-expressed recombinant cyclin/CDK1 and CK2 (NEB) as in our previous publication.Citation13 Roughly, 30 µl reaction mixtures with 3 µg of substrate protein (bait), 5 µM ATP, the kinase and either commercial (provided with kinases) or homemade kinase buffer (50 mM β-glycerophosphate, 20 mMNaF, 1.5 mM EGTA, 0.05 % NP-40 with 1 mM DTT, 1 mM PMSF, 500 µM Na3VO4 and supplemented with 10 mM MgCl2) were set and incubated at 30°C for 2 hours. In order to optimize the reactions, 8 μCi of radiolabeled [γ-32P] ATP was used and samples were run on SDS-PAGE, coomassie stained and analyzed with phosphoimager.
Cell culture and synchronization
HeLa cells were grown in Dulbecco modified Eagle's medium with 10% fetal bovine serum and penicillin-streptomycin. Cells were synchronized by double-thymidine method according to previously explained protocol.Citation13 Roughly, cells were blocked for 19 hrs with 2 mM thymidine and released into fresh medium for 9 hrs after washing with phosphate-buffered saline (PBS). Then, cells were blocked again with 2 mM thymidine for 16 hrs to arrest all the cells at the beginning of S phase. Finally, cells were washed with PBS and released into fresh medium. Synchronous progression of the cells through S, G2 and M phases were followed by flow cytometry analysis. Every two hours after the release, cells were collected and the cell cycle profiles were determined by propidium iodide staining followed by flow cytometry analysis (Fig. S1).
GST pull-downs
Cells were lysed in NP-40 Lysis Buffer (150 mM NaCl, 50 mM Tris-HCl (pH 8), 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1x protease inhibitor mixture (Roche) and 0.5% NP-40). 10 mg of whole cell extract were precleared with GSH beads by rocking for 2 hours at 4°C. 25 µg of phosphorylated GST-SLBPfragment, S/G2 stable mutant version or GST on beads were added into lysate and incubated at 4°C with end-over-end mixing for 2 hours. Baits were precipitated with GSH beads and washed 3 times with NP-40 Lysis Buffer for 30 minutes at 4°C. Proteins were eluted from GSH beads by adding 2X SDS Laemli Buffer and resolved on SDS-PAGE. The protein bands were stained using commercial Silver Stain for Mass Spectrometry kit (Pierce) according to manufacturer's protocol. Determined bands were cut and stored with 10 µl of methanol in order to prevent drying. LC-MS/MS analyses were performed in Institute of Biochemistry and Biophysics (Warsaw, Poland).
Transfection and RNAi interference
HeLa cells were seeded at 90% confluency on 6‐well plate and transfected with 2 µg DNA and Lipofectamine2000 (Invitrogen) according to manufacturer's protocol. Gene silencing was performed by transfecting HeLa cells with 50 nM siRNA ( GE Dharmacon) with DharmaFECT 1 (Dharmacon) according to the manufacturer's protocol. Cells were transfected with either ON-TARGETplus siGENOME Human DCAF11 siRNA or control ON-TARGETplus Non-Targeting pool (Dharmacon).
Immunoprecipitation
Cells were lysed in NP-40 Lysis Buffer. Around 1 mg of whole cell extract was incubated with 4 µg of indicated antibodies for 3–4 hrs at 4°C with shaking. Nonspecific IgG was used as a negative control. Immune complex was recovered on protein A sepharose beads by overnight rocking at 4°C. Precipitates were washed 3 times with the lysis buffer for 30 minutes and eluted by boiling in 2X SDS Laemli Buffer. Eluted proteins were resolved by SDS-polyacrylamide gel electrophoresis (PAGE), semi-dry transferred to a PVDF membrane and immunoblotted with indicated antibodies.
Analysis of cell death
Cell death in each culture was quantified using commercial colorimetric LDH toxicity kit (Roche) according to manufacturer's protocol. Absorbance density (OD) values at 480nm were measured using a plate reader.
Detection and quantification of BrdU incorporation
BrdU incorporation was detected using Colorimetric BrdU Detection Kit (Roche). In each culture, viable cell amount was also quantified using Wst-1 assay kit (Roche) and total BrdU incorporation levels were normalized to the viable cell numbers. In a combined protocol, cells were incubated in the presence of BrdU for 2 hours. Next, in order to quantify the viable cells, Wst- 1 reagent was added for 2 hours and OD values were measured at 420 nm with plate reader. After that, cells were carefully washed with PBS. Next, using Colorimetric BrdU Detection Kit (Roche), cells were fixed and stained with BrdU antibody according to manufacturer's protocol. BrdU incorporation values were quantified by OD measurements at 370 nm (reference wavelength: 492nm).
Drugs and antibodies
Cyclin A, SLBP (H-3) and Myc antibodies were purchased from Santa Cruz. DCAF11 (Sigma), HA (Covance), Skp1 (BD) antibodies were purchased from indicated companies. MG132 and Thymidine were purchased from Sigma.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Material.zip
Download Zip (1.2 MB)Acknowledgments
We thank Dr. Aziz Sancar, Dr. Nuri Öztürk, Dr. Yue Xiong, and Dr. Mathias Peter for kindly providing Cul4A and DCAF11 constructs.
Funding
This work was supported by The Scientific and Technological Research Council of Turkey (TUBITAK) grant # 110T987 (to MMK).
Related Research Data
References
- Singh RK, Liang D, Gajjalaiahvari UR, Kabbaj MH, Paik J, Gunjan A. Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle 2010; 9:4236-44; PMID:20948314; http://dx.doi.org/10.4161/cc.9.20.13636
- Ghule PN, Xie RL, Medina R, Colby JL, Jones SN, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Fidelity of histone gene regulation is obligatory for genome replication and stability. Mol Cell Biol 2014; 34:2650-9; PMID:24797072; http://dx.doi.org/10.1128/MCB.01567-13
- Ma Y, Kanakousaki K, Buttitta L. How the cell cycle impacts chromatin architecture and influences cell fate. Front Gen 2015; 6:19.
- Marzluff WF, Wagner EJ, Duronio RJ. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Gen 2008; 9:843-54; http://dx.doi.org/10.1038/nrg2438
- DeLisle AJ, Graves RA, Marzluff WF, Johnson LF. Regulation of histone mRNA production and stability in serum-stimulated mouse 3T6 fibroblasts. Mol Cell Biol 1983; 3:1920-9; PMID:6656760; http://dx.doi.org/10.1128/MCB.3.11.1920
- Heintz N, Sive HL, Roeder RG. Regulation of human histone gene expression: kinetics of accumulation and changes in the rate of synthesis and in the half-lives of individual histone mRNAs during the HeLa cell cycle. Mol Cell Biol 1983; 3:539-50; PMID:6406835; http://dx.doi.org/10.1128/MCB.3.4.539
- Harris ME, Bohni R, Schneiderman MH, Ramamurthy L, Schumperli D, Marzluff WF. Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Mol Cell Biol 1991; 11:2416-24; PMID:2017161; http://dx.doi.org/10.1128/MCB.11.5.2416
- Zheng L, Dominski Z, Yang XC, Elms P, Raska CS, Borchers CH, Marzluff WF. Phosphorylation of stem-loop binding protein (SLBP) on two threonines triggers degradation of SLBP, the sole cell cycle-regulated factor required for regulation of histone mRNA processing, at the end of S phase. Mol Cell Biol 2003; 23:1590-601; PMID:12588979; http://dx.doi.org/10.1128/MCB.23.5.1590-1601.2003
- Millevoi S, Vagner S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res 2010; 38:2757-74; PMID:20044349; http://dx.doi.org/10.1093/nar/gkp1176
- Rattray AM, Muller B. The control of histone gene expression. Biochem Soc Trans 2012; 40:880-5; PMID:22817752; http://dx.doi.org/10.1042/BST20120065
- Whitfield ML, Zheng LX, Baldwin A, Ohta T, Hurt MM, Marzluff WF. Stem-loop binding protein, the protein that binds the 3′ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol Cell Biol 2000; 20:4188-98; PMID:10825184; http://dx.doi.org/10.1128/MCB.20.12.4188-4198.2000
- Djakbarova U, Marzluff WF, Köseoğlu MM. Translation Regulation and Proteasome Mediated Degradation Cooperate to Keep Stem‐Loop Binding Protein Low in G1‐Phase. J Cell Biochem 2014; 115:523-30; PMID:24122909; http://dx.doi.org/10.1002/jcb.24686
- Koseoglu MM, Graves LM, Marzluff WF. Phosphorylation of threonine 61 by cyclin a/Cdk1 triggers degradation of stem-loop binding protein at the end of S phase. Mol Cell Biol 2008; 28:4469-79; PMID:18490441; http://dx.doi.org/10.1128/MCB.01416-07
- Koseoglu MM, Dong J, Marzluff WF. Coordinate regulation of histone mRNA metabolism and DNA replication: cyclin A/cdk1 is involved in inactivation of histone mRNA metabolism and DNA replication at the end of S phase. Cell cycle 2010; 9:3857-63; PMID:20935461; http://dx.doi.org/10.4161/cc.9.19.13300
- Enders GH. Mammalian interphase cdks: dispensable master regulators of the cell cycle. Gen Cancer 2012; 3:614-8; http://dx.doi.org/10.1177/1947601913479799
- Merrick KA, Fisher RP. Why minimal is not optimal: driving the mammalian cell cycle–and drug discovery–with a physiologic CDK control network. Cell cycle 2012; 11:2600-5; PMID:22732498; http://dx.doi.org/10.4161/cc.20758
- Ito S, Fujiyama-Nakamura S, Kimura S, Lim J, Kamoshida Y, Shiozaki-Sato Y, Sawatsubashi S, Suzuki E, Tanabe M, Ueda T, et al. Epigenetic silencing of core histone genes by HERS in Drosophila. Mol Cell 2012; 45:494-504; PMID:22365829; http://dx.doi.org/10.1016/j.molcel.2011.12.029
- Koseoglu MM, Graves LM, Marzluff WF. Cyclin A/Cdk1 and CK2 cooperate to trigger degradation of the Stem-loop binding protein (SLBP) at the end of S phase inhibiting histone mRNA biosynthesis. Febs J 2010; 277:143-.
- Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Gen Biol 2011; 12:220; http://dx.doi.org/10.1186/gb-2011-12-4-220
- He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev 2006; 20:2949-54; PMID:17079684; http://dx.doi.org/10.1101/gad.1483206
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell 2006; 23:709-21; PMID:16949367; http://dx.doi.org/10.1016/j.molcel.2006.08.010
- Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci 2009; 34:562-70; PMID:19818632; http://dx.doi.org/10.1016/j.tibs.2009.07.002
- Abbas T, Dutta A. CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle 2011; 10:241-9; PMID:21212733; http://dx.doi.org/10.4161/cc.10.2.14530
- Han J, Zhang H, Zhang H, Wang Z, Zhou H, Zhang Z. A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 2013; 155:817-29; PMID:24209620; http://dx.doi.org/10.1016/j.cell.2013.10.014
- Sharma P, Nag A. CUL4A ubiquitin ligase: a promising drug target for cancer and other human diseases. Open Biol 2014; 4:130217; PMID:24522884; http://dx.doi.org/10.1098/rsob.130217
- Huh J, Piwnica-Worms H. CRL4(CDT2) targets CHK1 for PCNA-independent destruction. Mol Cell Biol 2013; 33:213-26; PMID:23109433; http://dx.doi.org/10.1128/MCB.00847-12
- Shibutani ST, de la Cruz AF, Tran V, Turbyfill WJ, 3rd, Reis T, Edgar BA, Duronio RJ. Intrinsic negative cell cycle regulation provided by PIP box- and Cul4Cdt2-mediated destruction of E2f1 during S phase. Deve Cell 2008; 15:890-900; http://dx.doi.org/10.1016/j.devcel.2008.10.003
- Sullivan KD, Mullen TE, Marzluff WF, Wagner EJ. Knockdown of SLBP results in nuclear retention of histone mRNA. RNA 2009; 15:459-72; PMID:19155325; http://dx.doi.org/10.1261/rna.1205409
- Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol 2004; 6:1003-9; PMID:15448697; http://dx.doi.org/10.1038/ncb1172
- Wagner EJ, Berkow A, Marzluff WF. Expression of an RNAi-resistant SLBP restores proper S phase progression. Biochem Soc Trans 2005; 33:471-3; PMID:15916543; http://dx.doi.org/10.1042/BST0330471
- Mejlvang J, Feng Y, Alabert C, Neelsen KJ, Jasencakova Z, Zhao X, Lees M, Sandelin A, Pasero P, Lopes M, et al. New histone supply regulates replication fork speed and PCNA unloading. J Cell Biol 2014; 204:29-43; PMID:24379417; http://dx.doi.org/10.1083/jcb.201305017
- Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell 2010; 40:9-21; PMID:20932471; http://dx.doi.org/10.1016/j.molcel.2010.09.014
- Choe KP, Przybysz AJ, Strange K. The WD40 repeat protein WDR-23 functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in Caenorhabditis elegans. Mol Cell Biol 2009; 29:2704-15; PMID:19273594; http://dx.doi.org/10.1128/MCB.01811-08