
ABSTRACT
Multiple myeloma (MM) is still an incurable hematological malignancy. Despite recent progress due to new anti-myeloma agents, the pathology is characterized by a high frequency of de novo or acquired resistance. Delineating the mechanisms of MM resistance is essential for therapeutic advances. We previously showed that long-term genotoxic stress induces the establishment of a senescence-associated secretory phenotype, a pro-inflammatory response that favors the emergence of cells with cancer stem-like properties. Here, we studied the short-term response of MM cells following treatment with various DNA damaging agents such as the energetic C-ion irradiation. MM cells are highly resistant to all treatments and do not enter apoptosis after they arrest cycling at the G2 phase. Although the DNA damage response pathway was activated, DNA breaks remained chronically in damaged MM cells. We found, using a transcriptomic approach that RAD50, a major DNA repair gene was downregulated early after genotoxic stress. In two gerosuppression situations: induction of hypoxia and inhibition of the mammalian target of rapamycin (mTOR) pathway, we observed, after the treatment with a DNA damaging agent, a normalization of RAD50 expression concomitant with the absence of cell cycle arrest. We propose that combining inhibitors of mTOR with genotoxic agents could avoid MM cells to senesce and secrete pro-inflammatory factors responsible for cancer stem-like cell emergence and, in turn, relapse of MM patients.
Introduction
Multiple Myeloma (MM) is a hematological cancer characterized by the accumulation of clonal malignant plasma cells in the bone marrow leading to bone lytic lesions, anemia, and immunodeficiency.Citation1,2 The response rate and overall survival of MM patients have significantly improved due to new therapeutics such as immunomodulators, proteasome inhibitors, and autologous stem cell transplantation.Citation2,3 However, de novo or acquired resistance to therapy arises and is the major cause of lethality. MM resistance is thought to be due, at least in part, to the emergence of cancer stem-like cells (CSLCs).Citation4 Although the identity and functional properties of CSLCs are still debated, the existence of a sub-population of tumor initiating/propagating cells in MM is well documented.Citation5-12 CSLCs have been shown to be resistant to most therapeutics through both intrinsic properties and extrinsic mechanisms.Citation7,13-15 In a previous study, we showed that, following genotoxic stress such as X-ray irradiation or doxorubicin treatment, MM cancer cells enter senescence and release pro-inflammatory chemokines allowing the emergence of these CSLCs.Citation16 This is a long-term response.
It appeared from this study that targeting cancer cells before senescence onset might prevent emergence of MM CSLCs and in turn, resistance to therapeutics. We therefore investigated the short-term response of MM cells to genotoxic stresses in an attempt to understand early events and delineate a “therapeutic” window. To do so, we used carbon (C)-ion irradiation as a new genotoxic stress inducer on MM cells. C-ion irradiation has 2 main advantages over X-ray irradiation: a higher relative biological effectiveness and an increased linear energy transfer.Citation17 C-ion irradiation appeared as an interesting tool to generate numerous DNA double-strand breaks (DSBs) that are highly dependent on energy. However, we showed that MM cells are resistant to C-ion irradiations. Transcriptomic analysis validated by RT-PCR, revealed that the DNA repair gene RAD50 was downregulated upon C-ion irradiation. We further found that RAD50 was downregulated irrespective of genotoxic stress. Furthermore, although the DNA damage response (DDR pathway) was activated and the DNA repair program engaged, we observed ongoing DNA breaks causing the cell cycle arrest of MM cells. To prevent prolonged cell cycle arrest that would lead to senescence, we investigated the action of gerosuppressant agents on MM cells. We found that cell cycle arrest can be impeded by the induction of hypoxia, via the transcription factor hypoxia-inducible factor (HIF)-1α, or the inhibition of the mammalian target of rapamycin (mTOR) pathway using rapamycin. In addition, both gerosuppressant agents (HIF-1α and rapamycin) restored RAD50 gene expression. Our findings suggest that RAD50 is involved in gerosuppression and that rapamycin or rapamycin derivatives (i.e. rapalogs) could be envisaged for combination in anti-myeloma therapies.
Results
MM cells are resistant to energetic C-ion irradiations
C-ion irradiation is more energetic than X-ray irradiation.Citation17 As MM cells are resistant to X-ray irradiation (ref. 16 and Fig. S1A), we asked if C-ion irradiation could overcome MM cells resistance and triggers cell death. We exposed the RPMI 8226 MM cell line (hereafter referred to as 8226) to C-ion irradiation and followed cell viability over a 2-day period post-treatment. As control, we used the immortalized non-malignant B-lymphocyte CM cell line. Two days after a dose of 10 Gy of C-ion irradiation, CM cell viability dropped by 60% (25% of viability), while the viability of 8226 cells decreased by only 10% (80% of viability) (). A dose of 2 Gy was sufficient to significantly (p-value = 1.82 × 10−3 and 3.86 × 10−6, 24 h and 48 h post-irradiation, respectively) induce more cell death in CM cells than in 8226 cells after irradiation (Fig. S1B). Thus, in contrast to CM cells, 8226 cells were resistant to C-ion irradiation. We next compared the ability of C-ion and X-ray irradiations to induce cell death. As expected, the more energetic C-ion irradiation induced significantly (p-value = 2.92 × 10−4) more cell death than X-ray irradiation one day after a dose of 10 Gy but only in CM cells (). These data showed that 8226 cells were resistant to irradiations regardless of the source used at the doses tested.
Figure 1. MM cells are resistant to C-ion and X-ray irradiations (A) Exponentially growing CM and 8226 cells were irradiated with the indicated doses of C-ion (2–10 Gy) or left non-irradiated as controls (0 Gy) and the percentage of cell viability evaluated by Trypan blue exclusion 24 and 48 h post-treatment. The graphs represent the percentage of cell viability (CM cells, left graph; 8226 cells, right graph) as a function of the dose of irradiation and as a function of time. The experiment has been carried out 3 times in duplicate or triplicate (B) CM and 8226 cells were cultured as before but cells were irradiated either by C-ion or X-rays at the doses indicated and the percentage of cell viability evaluated one day later. The bar graph illustrates the percentage of CM (left graph) and 8226 (right graph) cells viability as a function of irradiation doses. The experiment has been carried out 3 times in duplicate or triplicate (C) CM and 8226 cells were cultured as before and irradiated either by C-ion or X-rays at the doses indicated (2 or 10 Gy) and the OTM evaluated one day later by the alkaline comet assay. OTMs obtained at 0 Gy were subtracted from those obtained at 2 or 10 Gy and OTMs were subsequently averaged. The graphs represent the mean of OTM as a function of the dose of irradiations. The left graph depicts the mean OTM of 8226 and CM cells at 2 and 10 Gy C-ion irradiations, whereas the right graph shows the mean OTM of 8226 cells after either X-rays or C-ion irradiations. **, p< 0.01 with the t-test.
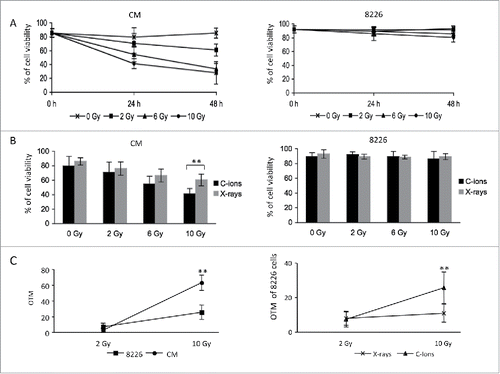
Irradiations cause DNA double-strand breaks (DSBs); if damage is overwhelming cells die or senesce.Citation18 Using the comet assay, DSBs formation was investigated one hour after C-ion irradiation. A dose of 10 Gy increased the mean of the olive tail moment (OTM) of CM cells significantly more than that of 8226 cells (p-value = 5.53 × 10−11). This demonstrated that C-ion irradiation induced more DNA breaks in CM cells than in 8226 cells, underlying CM sensitivity (, left panel). We then compared DNA breaks in 8226 cells one hour after C-ion or X-ray irradiation (, right panel). Significantly more DNA breaks were induced in 8226 cells by C-ion irradiation when compared to X-rays (p-value = 1.63 × 10−6). Thus, in contrast to CM cells, the more energetic C-ion irradiation did not induce more cell death in 8226 cells, although more DNA breaks were detected. This indicated that using a more energetic source of irradiation did not overcome initial MM cells resistance. Furthermore, the mechanisms of resistance of MM cells to irradiations or even to DNA damaging agents could be similar.
C-ion irradiation leads to the recruitment of DNA damage/telomere stress-induced senescence pathway
To understand the initial mechanisms of resistance of MM cells to irradiation, we analyzed the genome-wide gene expression of C-ion-irradiated 8226 cells using microarrays. Microarray data and annotations have been deposed in the NIH Gene Expression Omnibus under accession number GSE76471. In this short-term response, a total of 1,745 genes were found to be differentially expressed in 8226 cells one day after a 6 Gy dose of C-ion irradiation when compared to non-irradiated cells (p-value = 0.051). Real-time RT-PCR was performed for validation of microarray results. We found a strong correlation between microarray and RT-PCR data for the expression of CEP110, MSH5 and SOX2 genes (Fig. S2A). Thereafter, this list of genes was implemented by the Reactome Pathway Browser (http://www.reactome.org/PathwayBrowser/). Five pathways were statistically overrepresented among these 1,745 genes (false discovery rate (FDR) < 0.051) (). The identified pathways fell under categories of cellular processes associated with cell senescence and its effector programs. Indeed, telomerase shortening and genomic damage through the activation of the DDR pathway are involved in both the induction and maintenance of senescence.Citation19 Drastic changes in chromatin structure are associated with cellular senescence and nucleolar repression facilitates initiation and maintenance of senescence. Epigenetic changes accompany senescence but are also able to initiate the process in particular through the induction of histone deacetylases (HDACs).Citation20 Moreover, an efficient regulation of rRNA transcription controlled by RNA Polymerase I is necessary for senescence.Citation21 Sirtuin 1 (or SIRT1) is a class of HDAC that promotes proliferation and prevents senescence.Citation22,23 These data showed that one day after the genotoxic stress induced by C-ion irradiation, the main effector programs of senescence were recruited.
Table 1. Pathways overrepresented among the 1,745 genes differentially expressed identified by the Reactome pathway browser.
Genotoxic stresses lead to a down-regulation of the DNA repair gene RAD50 in MM cells
In the overrepresented DNA damage/telomere senescence pathway, 26 genes among the 1,745 initially differentially expressed genes were isolated. Those genes code for several kinds of histone (19/26) and proteins with various roles within the stress/telomere senescence program. The DNA repair gene RAD50 was among these 26 genes. This gene was of interest as its downregulation would lead to either a default of DDR pathway activation and/or a lack in DNA repair. Indeed, following DSBs, DNA lesions are recognized by sensor proteins of the MRN complex which consists of meiotic recombination 11 (MRE11), RAD50 and Nijmegen breakage syndrome 1 (NBS1) proteins. The detection of DNA damage is followed by the recruitment of signaling and repair proteins and alteration of chromatin structures including histone modifications to permit access to DNA. Apical kinases of the DDR pathway including ATM, are subsequently activated to ensure cell response. We verified that DDR was activated following X-ray irradiation. This was the case since both checkpoint kinases 1 and 2 (CHK1/2) were phosphorylated one hour post-irradiation. CHK2 phosphorylation was sustained 24 hours post-irradiation (Fig. S2B).
In addition, the MRN complex regulates DSBs repair through the homologous recombination (HR) or non-homologous end-joining (NHEJ) processes.Citation24 RT-PCR analysis confirmed the downregulation of RAD50 gene expression one day after a 6 Gy dose of C-ion irradiation (). Furthermore, we showed that RAD50 gene expression was downregulated also one day after X-ray irradiation or doxorubicin treatment (). In addition, RAD50 was the only member of the MRN complex to be downregulated one day after X-ray irradiation (Fig. S2C). This indicated that downregulation of RAD50 gene expression in 8226 cells occurred after several types of genotoxic stresses.
Figure 2. RAD50 gene expression is downregulated in response to DNA damaging agents while cells are arrested at the G2/M phase (A) RAD50 expression was analysis using RT-PCR in 8226 cells one day after a dose of 6 Gy C-ion or X-ray irradiation, or one day after a treatment with 25 nM doxorubicin. Results are presented as the relative expression (in arbitrary units, A.U.) of RAD50 gene in stressed 8226 cells normalized to internal control gene (GAPDH) and relative to unstressed cells normalized to 1. The experiment has been carried out 3 times in triplicate. Data are mean ± SD (B) 8226 cells were subjected to a 10 Gy dose of C-ion irradiation. Cells were harvested and analyzed by the alkaline comet assay or stained with an antibody anti-γH2AX followed by cytometry analysis. OTMs were calculated at different time points (1, 24 and 72 h). On the top are represented the means of OTM values as well as SD as a function of time after irradiation. Values were calculated after subtracting background (OTMs in non-irradiated cells) from 3 independent experiments carried out in triplicate. Representative comet images from one experiment illustrate this experiment. Below, residual γH2AX level was quantified as the ratio signal post-irradiation vs. signal in non-irradiated cells. This ratio relative to the DNA content was plotted on the graph. The experiment has been done once and is illustrated by the corresponding cytometric profiles (C) 8226 cells were irradiated with C-ion at the dose indicated (2-10 Gy) and harvested one day later. DNA content was measured by flow cytometry. The graph illustrates the percentage of cells in the different phases of the cell cycle as a function of the irradiation doses. Data are mean as well as SD from 3 independent experiments carried out in triplicate.
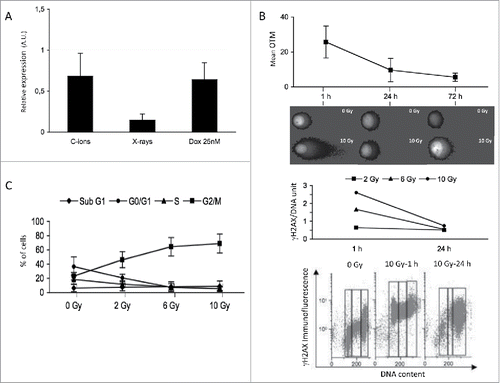
To evaluate the ability of 8226 cells to repair DNA breaks, OTMs were determined 1 d and 3 d after C-ion irradiation. While most of the DNA breaks were repaired at day one, some DNA breaks remained until day 3 (). Similar results were obtained using γH2AX staining after C-ion () or X-ray irradiation (Fig. S1C). These results suggested that the remaining DNA breaks resulted most probably from a defect in the dynamics of the MRN complex caused by the downregulation of RAD50 gene expression upon genotoxic stress. To investigate if these remaining DNA breaks were able to activate cell cycle checkpoints, the cell cycle profile of irradiated cells was established. The cell cycle distribution of 8226 cells was evaluated after propidium iodide (PI) incorporation and cytometry analysis. It appeared that the percentage of cells in the G2/M phase was enriched one day after a dose of 6 Gy of C-ion irradiation (). Similar results were obtained after X-ray irradiation (Fig. S2D). These data suggested that, in the short term, the remaining DNA breaks were able to perturb cellular functioning of MM cells by arresting the cell cycle at the G2/M phase.
Cell cycle arrest can be overcome by HIF-1α induction
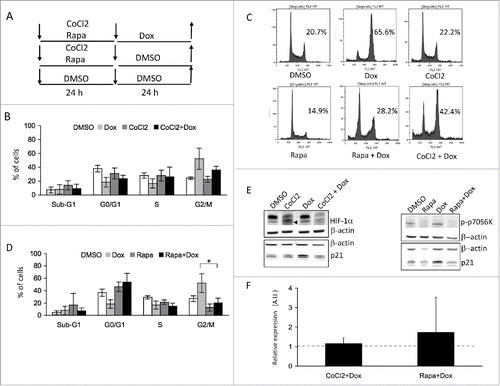
MM cells niche in the hypoxic bone marrow environment.Citation25 Under hypoxic conditions, cells express the transcription factor HIF-1α, which has been shown to sensitize cells to genotoxic stress.Citation26 We tested if HIF-1α expression could overcome genotoxic stress resistance in 8226 cells. To do so, 8226 cells were treated with CoCl2 that mimics hypoxia, before a doxorubicin treatment as DSBs inducer (). While doxorubicin led to a cell cycle arrest one day after treatment, a pretreatment with CoCl2 prevented MM cells from this arrest (). Western blot analysis confirmed the induction of hypoxia by CoCl2 with the increase of HIF-1α (, left panel). Moreover, while the cell cycle inhibitor p21 was increased in arrested doxorubicin-treated 8226 cells, p21 decreased in doxorubicin-treated cells under hypoxic conditions. The decrease of p21 level mirrored the absence of cell cycle arrest. This showed that under hypoxic conditions, MM cells did not arrest upon genotoxic stress.
Figure 3. HIF-1α and rapamycin act as gerosuppressants in MM cells treated with doxorubicin (A) Schematic representation of treatments for cultured cells. 8226 cells were seeded at day 0, then pre-treated with CoCl2 (300 μM), rapamycin (Rapa, 40 nM) or DMSO (0.01% as control) for 24 h. Cells were then washed twice and seeded in new medium containing either DMSO (0.01% as control) or treated with 25 nM doxorubicin (B) 8226 cells were treated with vehicle (DMSO), pretreated with CoCl2 for 24 h (CoCl2) then treated with doxorubicin (CoCl2+Dox) or non-pretreated (Dox). Cells were then assessed for DNA content after PI staining and cytometry analysis. Experiments have been repeated 3 times. At least 10,000 events were gated for each experimental condition (C) Representative experiments showing cytometry profiles is shown. On the graphs, are indicated the percentage of cells in the G2/M phase of the cell cycle, under the graph is indicated the treatment (D) 8226 cells were treated with vehicle (DMSO), pretreated with rapamycin for 24 h (Rapa) then treated with doxorubicin (Rapa+Dox) or non-pretreated (Dox). Cells were then assessed for DNA content after PI staining and cytometry analysis. Percentages of cells within the various phases of the cell cycle are indicated (mean ± SD) on the graph. Experiments have been repeated 3 times. At least 10,000 events were gated for each experimental condition. * p < 0.05 with the t-test (E) Western blots illustrate the protein levels of HIF-1α (the specific band is marked by an arrow), p-p70S6K, p21 and β-actin (as control) under the various conditions of culture (F) RT-PCR analysis of RAD50 gene expression one day after rapamycin (40 nM) or CoCl2 (300 μM) pretreatment followed by doxorubicin (25 nM) treatment. Results are presented as the relative expression (in A.U.) of RAD50 gene in 8226 cells normalized to internal control gene (GAPDH) and relative to DMSO-treated cells normalized to 1. The experiment has been carried out 3 times in triplicate.
RAD50 downregulation and cell cycle arrest are controlled by a HIF1α/mTOR axis
Our previous work showed that genotoxic stresses trigger, as a long-term response, a SASP and emergence of CSLCs.Citation16 Moreover, by suppressing geroconversion, hypoxia prevents senescence by inhibiting geroconversion and, hence, tumorigenesis.Citation27 Leontieva and collaborators have demonstrated that hypoxic conditions caused gerosuppression via inhibition of the mTOR pathway in non-malignant cells.Citation28,29 Therefore, we analyzed the cell cycle distribution in 8226 cells pretreated, one day before doxorubicin treatment, with rapamycin to inhibit mTOR (). Rapamycin pretreatment abrogated significantly (p-value = 0.048) the cell cycle arrest caused by a 24 h-treatment with doxorubicin (). Western blot analysis confirmed mTOR inhibition as its target, p70S6K was not phosphorylated upon rapamycin addition (, right panel). Moreover, this analysis indicated that p21 protein level increased upon doxorubicin treatment and decreased after rapamycin pretreatment (). This confirmed, at a molecular level, the absence of cell cycle arrest. Our data showed that rapamycin can act as gerosuppressant in malignant MM cells.
We next tested the action of both gerosuppressants, HIF1α and rapamycin on NCI-H929 MM cells. These cells are TP53 wild-type whereas 8226 cells are TP53 mutated. No cell cycle arrest was observed after genotoxic stress when cells were pretreated with either of the 2 gerosuppressants (Fig. S3A-C). Western blot analysis confirmed the induction of hypoxia by CoCl2, mTOR inhibition upon rapamycin treatment and decreased p21 protein level upon pretreatment with gerosuppressants (Fig. S3D). This suggests that the genomic status of TP53 is not essential. However, we observed that NCI-H929 cells were more sensitive to rapamycin than 8226 cells in the G0-G1 phase, perhaps reflecting their differences in the regulation of cyclin D1.Citation30,31 As cell cycle block was concomitant with RAD50 expression downregulation, RAD50 expression was examined one day after treatment of 8226 cells with either rapamycin or CoCl2. Hence, with both gerosuppressants, RAD50 expression reached a level comparable to the one in untreated 8226 cells ().
Discussion
In this study, we analyzed, for the first time, the response of MM cells to C-ion irradiation. As MM cells are resistant to X-rays, we thought that more energetic C-ion could induce MM cell death. C-ion irradiation induced more DNA breaks in MM cells when compared to X-ray irradiation. However, these additional DNA breaks did not cause more cell death, showing that MM cells are also resistant to C-ion irradiation. Interestingly, monitoring the evolution of these DNA breaks over time revealed that some breaks remained 3 d post-irradiation. Hence, ongoing DNA breaks and not the number of DNA breaks per se conditioned MM cell fate after irradiation.
DNA breaks can be repaired via 2 main mechanisms: NHEJ and HR. NHEJ is thought to be error prone, while HR is considered to be error-free. MM is characterized by a high genomic instability, driving force of tumorigenesis. Previous studies have pointed to an imbalance of DNA repair mechanisms toward HR in MM cells.Citation32 This imbalance is inherited due to genetic polymorphism and/or gene expression deregulation. Our transcriptomic analysis revealed that RAD50 gene expression is downregulated upon C-ion irradiation as well as upon X-ray irradiation or doxorubicin treatment. RAD50 participates in DNA repair within the HR as part of the MRN complex with MRE11 and NBS1. Of note, RAD50 gene was the only member of the MRN complex to be downregulated following X-ray irradiations. Proteins of the MRN complex recognize DNA breaks allowing the activation of ATM. Activated ATM orchestrates the DDR leading, among others, to DNA repair.Citation33 Thus, downregulation of RAD50 expression, upon genotoxic stress, might impair HR function in MM cells. NHEJ being intrinsically weakened in MM,Citation32 further impairment of HR could explain observed ongoing DNA breaks. These results indicate that genotoxic stress is able to affect the inherited imbalance between NHEJ and HR in MM, causing unrepaired DNA breaks and programming senescence.
Deregulated DSBs repair in MM cells has been linked also to a high expression of the recombinase RAD51 that is involved in the HR process. This high expression is concomitant with an upregulation of HR activity.Citation34 RAD51 gene expression was not altered in our transcriptomic study. RAD51 has been shown to interact with cyclin D1 in cancer cells over-expressing cyclin D1.Citation35 We confirmed this interaction in MM cells as revealed by high throughput proteomic screen for cyclin D1 protein partners in the U266 MM cell line which expresses high level of cyclin D1 (unpublished data). Although present in 8226 cells, cyclin D1 level is low.Citation36 Therefore the RAD50 pathway may be preferred to the RAD51 one. Although cyclin D1 is present in 40-50% of MM patients, a high cyclin D1 expression is linked to the presence of t(11;14)(q13;q32) found in only 15% of MM cases.Citation37 This is in favor of an important role of RAD50.
Transcriptomic data revealed that apart of the DDR and telomere stress-induced senescence/packaging telomere ends pathways, genotoxic stress recruits RNA Pol I promoter opening, HDAC and SIRT1-controlled rRNA expression pathways (). A hallmark of cellular senescence is change in the cell morphology, metabolism, gene expression, and the increased incidence of senescence-associated heterochromatin foci, and other modification of chromatin structures in the nucleus.Citation38,39 These chromatin modifications include histone variants, histone modifications, but more generally, global epigenetic changes allowing chromatin accessibility in senescent cells. Histone deacetylation has a pivotal role in the regulation of epigenetic events leading to transcription silencing. HDACs modulate the DDR cascade at various levels. On the one hand, HDAC inhibitors amplify DNA damage through the production of reactive oxygen species and the degradation of DDR effectors, and on the other hand, they reduce DNA repair leading to cell death. SIRT1, a type III HDAC, was also shown to modulate DNA damage-induced cell death. But importantly, HDAC and SIRT1 are recognized as senescence modulators. SIRT1 elicits its effects by regulating the acetylation status of a plethora of targets involved in senescence such as p53, NF-κB, mTOR and the FOXO family of transcription factors.Citation40 As a whole the initiation of senescence in injured MM cells in response to DNA damage, requires a bona fide senescence network.
MM cells enter senescence 7 d post X-ray irradiations or 2 d post-doxorubicin treatment.Citation16 Senescence is characterized by an irreversible cell cycle arrest while sustaining cellular growth. This uncoupling of cell proliferation and cellular growth thus unbalances cellular metabolism. Actually, senescent cells might maintain an apparent homeostasis by eliminating oversynthesized factors such as those observed in the SASP. SASP has been shown to contribute to the pro-tumorigenic effect of senescence,Citation41,42 including in MM.Citation16 Therefore, preventing SASP in MM might lead to a decreased tumorigenesis. Among the roles of mTOR is the control of cellular metabolism, and hence mTOR has been shown to drive senescence entry.Citation43 Here, we showed that mTOR inhibition via rapamycin impedes cell cycle arrest induced by genotoxic stresses, thus preventing MM cells to enter senescence. Interestingly, this effect was achieved with a pretreatment. Cells did not require rapamycin while being subjected to genotoxic stresses. This suggests that mTOR plays a pivotal role in driving geroconversion in MM cells.
mTOR pathway is constitutively active in MM cells.Citation44 mTOR is part of 2 complexes: mTORC1 and mTORC2. Because rapamycin preferentially inhibits mTORC1, it would be interesting to test if INK128, which inhibits both complexes, also acts as gerosuppressant. In this study, hypoxia induction also led to the absence of cell cycle arrest after genotoxic stress. Both gerosuppressants were active independently of TP53 status. Rapamycin and hypoxia reflect a change in cellular metabolism synonym of decreased biogenesis; and hence, both act as gerosuppressant. Together, these results suggest that mTOR inhibitors and HIF-1α inducers might prevent and/or delay the emergence of CSLCs and therefore the refueling of MM tumors, but they would not prevent the proliferation of non MM CSLCs.
Senescence can not only be induced by unrepaired DNA damages but also by shortened telomeres.Citation45 Interestingly, RAD50 has been shown to participate in telomere length maintenance, and if disrupted to lead to short telomere and senescence in Arabidopsis and in yeast.Citation46,47 Here, we showed that RAD50 gene expression is restored upon rapamycin or hypoxia pretreatment in doxorubicin-treated cells. Furthermore, telomerase being active in MM cells,Citation48 these results point to a similar role of RAD50 in driving senescence, whether it is caused by shorter telomere or unrepaired DNA damages. Halicka et al. have shown that gerosuppressive agents, including rapamycin, reduced the level of expression of γH2AX, in various cell types.Citation49 It is of interest to examine if RAD50 is involved in this decreased expression of γH2AX. DNA damages are able to decelerate, and even arrest, cell cycle so that they can be repaired. Thus, it is not surprising that RAD50, which participates in DNA repair, could modulate cell entry in the irreversible arrest of senescence.
Material and methods
Cell culture and treatment
8226, NCI-H929 and CM cells have been described previously.Citation16 Cells were cultured at 37°C under 5% CO2 in RPMI 1640 medium (Lonza SPRL) supplemented with 10% fetal calf serum (FCS, PAA Laboratories GmbH), 2 mM L-glutamine and antibiotics (Lonza). Cell viability was followed using Trypan blue exclusion tests.
To induce genotoxic stress, cells were either irradiated or treated with different concentrations of doxorubicin as indicated in figures' legend. For irradiation experiments, cells were either irradiated with C-ion on the D1 line of the GANIL accelerator (Caen, France; 75 MeV/n, 1 Gy/min) or with X-rays using an Orion generator (CGR MEV, Riverside, CA, USA; 5 MeV, 1.4 Gy/min) or a Saturne 15 (Siemens, 15MeV, 6 mA, 2 Gy/min). C-ion runs were carried out between 2009 and 2011.
Alkaline comet assay
Alkaline comet assay was performed as described by Laurent et al.Citation50 Comet images were taken using a confocal microscope (Olympus FV1000). Images were analyzed using the computer image analysis software Casp to obtained the Olive Tail Moment (OTM). At least 100 images per sample and experiment were analyzed. OTMs obtained at 0 Gy were subtracted from those obtained at 2 or 10 Gy and OTMs were subsequently averaged.
Flow cytometry analysis
For cell cycle experiments, cells were washed with PBS and fixed in 70% ethanol /PBS at −20°C for at least 30 min. Cells were then washed with PBS and incubated in PBS containing 10 μg/ml of propidium iodide (PI) and 100 μg/ml of RNAse A. On average, 104 cells were analyzed on either an Epics XL or Gallios cytometer and data were processed with the Expo 32™ and Kaluza softwares, respectively (Beckman Coulter).
For cytometry analysis of γH2AX, 8226 cultured cells were C-ion or X-ray irradiated (2–10 Gy) and harvested 1 or 24 h later. Cells were fixed in 70% ethanol/PBS, washed in PBS and permeabilized in 0.25% Triton X100/PBS. Staining was performed by incubation for 2 h with anti-γH2AX antibody diluted 1/1,000 in PBS containing 1% bovine serum albumin (clone JBW301, #05-365, Millipore). Cells were then counterstained with PI/RNase A and analyzed. Phosphorylation of H2AX per DNA content was measured. Data were normalized so that γH2AX signal was independent of the cell cycle phase. To do so, mean of γH2AX intensities in S and G2/M were divided by 1.5 and 2, respectively. Each experiment has been conducted once.
Western blotting
Western blotting was performed as previously described in Cahu et al.Citation16 The following antibodies were used: anti-HIF-1α (sc-10790) and anti-β-actin (sc-4778) from Santa Cruz Biotech. (Santa Cruz, CA); anti-p21CIP1 (#2946), anti-CHK1 (#2360), anti-CHK2 (#6334), anti-phospho(p)Thr389-p70S6K (#9205), anti-pSer345-CHK1 (#2341), and anti-pThr68-Chk2 (#2197) from Cell Signaling Tech. (Danvers, MA). The anti-β-actin antibody served for loading control.
Quantitative RT-PCR
Total RNA was extracted using Trizol reagent and cDNAs were generated using M-MLV reverse transcriptase according to the manufacturer instructions (Life Technologies SAS). QRT-PCRs were performed using SYBR Green Master Mix with a SDS7000 apparatus (Applied Biosystems Inc.) according to the manufacturers' instructions. Each reaction condition was performed in triplicate. The entire experiment has been repeated 3 times with RNAs extracted from different cell culture batches. Standard curves were generated for each gene to determine PCR efficiencies, and relative gene expression was calculated by the 2−ΔΔCt method with GAPDH gene as internal control for normalization according to Schmittgen and Livak.Citation51 The sequences of primers used in the present study are presented in the supplementary Table S1.
Microarrays
Total RNA was extracted using Trizol reagent and cDNAs were generated using M-MLV reverse transcriptase according to the manufacturer instructions (Life Technologies SAS). Biotinylated single strand cDNA targets were prepared, starting from 250 ng of total RNA, using the Ambion WT Expression Kit (#4411974) and the Affymetrix GeneChip® WT Terminal Labeling Kit (#900671) according to Affymetrix recommendations. Following fragmentation and end-labeling, 1.9 μg of cDNAs were hybridized for 16 h at 45°C on GeneChip® Human Gene 1.0 ST arrays (Affymetrix) interrogating 28,869 genes represented by approximately 27 probes spread across the full length of the gene. The chips were washed and stained in the GeneChip® Fluidics Station 450 (Affymetrix) and scanned with the GeneChip® Scanner 3000 7G (Affymetrix) at a resolution of 0.7 μm. Raw data (.CEL Intensity files) were extracted from the scanned images using the Affymetrix GeneChip® Command Console (AGCC) version 2.0., CEL files were further processed with Affymetrix Expression Console software version 1.1 to calculate probe set signal intensities using Robust Multi-array Average (RMA) algorithms with default settings.
Statistical significance of individual gene expression levels was analyzed using a in house R script. Significant genes were identified based on fold-changes (FC). All data have been submitted to Gene Expression Omnibus database repository (accession number: GSE76471).
Statistical analyses
Student's t test was used to determine the significance of differences between experimental groups. Data were analyzed with 2-tailed tests and p < 0.05 was considered significant.
Abbreviations
| ATM | = | ataxia telangiectasia mutated protein |
| C | = | carbon |
| CHK1/2 | = | checkpoint kinase 1/2 |
| CSLC | = | cancer stem-like cell |
| DDR | = | DNA damage response |
| DSB | = | double-strand break |
| FDR | = | false discovery rate |
| HDAC | = | histone deacetylase |
| HIF-1α | = | hypoxia-inducible factor-1α |
| HR | = | homologous recombination |
| MM | = | multiple myeloma |
| MRE11 | = | meiotic recombination 11 |
| mTOR | = | mammalian target of rapamycin |
| NBS | = | Nijmegen breakage syndrome 1 |
| NHEJ | = | non-homologous end-joining |
| p- | = | phosphorylated |
| OTM | = | olive tail moment |
| PI | = | propidium iodide |
| SD | = | standard deviation |
| SIRT | = | sirtuin |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1196302_Supplemental_Material.zip
Download Zip (1.4 MB)Acknowledgments
We thank Anne Barbaras for technical assistance, and the technical platform of cytometry of SFR ICORE (Université de Caen Normandie, France). The transcriptomic data have been obtained thanks to I.G.B.M.C. microarray and sequencing platform (Strasbourg, France).
Funding
This work received financial supports from Ligue contre le Cancer-Comité du Calvados (to BS), and Conseil Régional de Basse-Normandie and Naturalia & Biologia (to JC).
Related Research Data
References
- Gozzetti A, Candi V, Papini G, Bocchia M. Therapeutic advancements in multiple myeloma. Front Oncol 2014; 4:241-5; PMID:25237651; http://dx.doi.org/10.3389/fonc.2014.00241
- Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet 2015; 385:2197-208; PMID:25540889; http://dx.doi.org/10.1016/S0140-6736(14)60493-1
- Mimura N, Hideshima T, Anderson KC. Novel therapeutic strategies for multiple myeloma. Exp Hematol 2015; 43:732-41; PMID:26118499; http://dx.doi.org/10.1016/j.exphem.2015.04.010
- Franqui-Machin R, Wendlandt EB, Janz S, Zhan F, Tricot G. Cancer stem cells are the cause of drug resistance in multiple myeloma: fact or fiction? Oncotarget 2015; 6:40496-506; PMID:26415231
- Pilarski L, Seeberger K, Coupland R, Eshpeter A, Keats J, Taylor B, Belch AR. Leukemic B cells clonally identical to myeloma plasma cells are myelomagenic in NOD/SCID mice. Exp Hematol 2002; 30:221-8; PMID:11882359; http://dx.doi.org/10.1016/S0301-472X(01)00788-3
- Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, Smith BD, Civin CI, Jones RJ. Characterization of clonogenic multiple myeloma cells. Blood 2004; 103:2332-6; PMID:14630803; http://dx.doi.org/10.1182/blood-2003-09-3064
- Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, McNiece I, Lin L, Ambinder RF, Peacock C, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res 2008; 68:190-7; PMID:18172311; http://dx.doi.org/10.1158/0008-5472.CAN-07-3096
- Reghunathan R, Bi C, Liu SC, Loong KE, Chung TH, Huang G, Chng WJ. Clonogenic multiple myeloma cells have shared stemness signature associated with patient survival. Oncotarget 2013; 4(8):1230-40; PMID:23985559; http://dx.doi.org/10.18632/oncotarget.1145
- Zhou W, Yang Y, Gu Z, Wang H, Xia J, Wu X, Zhan X, Levasseur D, Zhou Y, Janz S, et al. ALDH1 activity identifies tumor-initiating cells and links to chromosomal instability signatures in multiple myeloma. Leukemia 2014; 28:1155-8; PMID:24365790; http://dx.doi.org/10.1038/leu.2013.383
- Chen Z, Orlowski RZ, Wang M, Kwak L, McCarty N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood 2014; 123:2204-8; PMID:24425802; http://dx.doi.org/10.1182/blood-2013-07-517136
- Hosen N, Matsuoka Y, Kishida S, Nakata J, Mizutani Y, Hasegawa K, Mugitani A, Ichihara H, Aoyama Y, Nishida S, et al. CD138-negative clonogenic cells are plasma cells but not B cells in some multiple myeloma patients. Leukemia 2012; 26:2135-41; PMID:22430638; http://dx.doi.org/10.1038/leu.2012.80
- Kim D, Park CY, Medeiros BC, Weissman IL. CD19-CD45low/-CD38hig/CD138+plasma cells enrich for human tumorigenic myeloma cells. Leukemia 2012; 26:2530-7; PMID:22733078; http://dx.doi.org/10.1038/leu.2012.140
- Jakubikova J, Adamia S, Kost-Alimova M, Klippel S, Cervi D, Daley JF, Cholujova D, Kong SY, Leiba M, Blotta S, et al. Lenalidomide targets clonogenic side population in multiple myeloma: pathophysiologic and clinical implications. Blood 2011; 117:4409-19; PMID:21321360; http://dx.doi.org/10.1182/blood-2010-02-267344
- Chaidos A, Barnes CP, Cowan G, May PC, Melo V, Hatjiharissi E, Papaioannou M, Harrington H, Doolittle H, Terpos E, et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013; 121:318-28; PMID:23169779; http://dx.doi.org/10.1182/blood-2012-06-436220
- Van Valckenborgh E, Matsui W, Agarwal P, Lub S, Dehui X, De Bruyne E, Menu E, Empsen C, van Grunsven L, Agarwal J, et al. Tumor-initiating capacity of CD138- and CD138+ tumor cells in the 5T33 multiple myeloma model. Leukemia 2012; 26:1436-9; PMID:22289925; http://dx.doi.org/10.1038/leu.2011.373
- Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis 2012; 3:e446; PMID:23254289; http://dx.doi.org/10.1038/cddis.2012.183
- Schlaff CD, Krauze A, Belard A, Connell JJO, Camphausen KA. Bringing the heavy: carbon ion therapy in the radiobiological and clinical context. Radiat Oncol 2014; 9:88; PMID:24679134; http://dx.doi.org/10.1186/1748-717X-9-88
- Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 2012; 12:801-17; PMID:23175119; http://dx.doi.org/10.1038/nrc3399
- Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol 2013; 75:685-705; PMID:23140366; http://dx.doi.org/10.1146/annurev-physiol-030212-183653
- Garcia SN, Pereira-Smith O. MRGing chromatin dynamics and cellular senescence. Cell Biochem Biophys 2008; 50:133-41; PMID:18231726; http://dx.doi.org/10.1007/s12013-008-9006-7
- Yang L, Song T, Chen L, Chen HSJ. Nucleolar repression facilitates initiation and maintenance of senescence. Cell Cycle 2015; 14:3613-23; PMID:26505814; http://dx.doi.org/10.1080/15384101.2015.1100777
- Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer 2009; 9:123-8; PMID:19132007; http://dx.doi.org/10.1038/nrc2562
- Kim EJ, Um SJ. SIRT1: roles in aging and cancer. BMB Reports 2008; 41:751-6; PMID:19017485; http://dx.doi.org/10.5483/BMBRep.2008.41.11.751
- Stracker TH, Petrini JHJ. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 2011; 12:90-103; PMID:21252998; http://dx.doi.org/10.1038/nrm3047
- Martin SK, Diamond P, Gronthos S, Peet DJ, Zannettino ACW. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia 2011; 25:1533-42; PMID:21637285; http://dx.doi.org/10.1038/leu.2011.122
- Moeller BJ, Dreher MR, Rabbani ZN, Schroeder T, Cao Y, Li CY, Dewhirst MW. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell 2005; 8:99-110; PMID:16098463; http://dx.doi.org/10.1016/j.ccr.2005.06.016
- Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci USA 2012; 109:13314-8; PMID:22847439; http://dx.doi.org/10.1073/pnas.1205690109
- Leontieva OV, Blagosklonny MV. Hypoxia and gerosuppression: the mTOR saga continues. Cell Cycle 2012; 11:3926-31; PMID:22987149; http://dx.doi.org/10.4161/cc.21908
- Leontieva OV, Demidenko ZN, Blagosklonny MV. S6K in geroconversion. Cell Cycle 2013; 12:3063-9; PMID:23974099; http://dx.doi.org/10.4161/cc.26130
- Strömberg T, Dimberg A, Hammarberg A, Carlson K, Österborg A, Nilsson K, Jernberg-Wiklund H. Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood 2004; 103:3138-3147; PMID:15070696; http://dx.doi.org/10.1182/blood-2003-05-1543
- Frost P, Shi Y, Hoang B, Gera J, Lichenstein A. Regulation of D-cyclin translation inhibition in myeloma cells treated with mammalian target of rapamycin inhibitors: rationale for combined treatment with extracellular signal-regulated kinase inhibitors and rapamycin. Mol Cancer Ther 2009; 8:83-93; PMID:19139116; http://dx.doi.org/10.1158/1535-7163.MCT-08-0254
- Gourzones-Dmitriev C, Kassambara A, Sahota S, Rème T, Moreaux J, Bourquard P, Hose D, Pasero P, Constantinou A, Klein B. DNA repair pathways in human multiple myeloma. Cell Cycle 2014; 12:2760-73; http://dx.doi.org/10.4161/cc.25951
- Cremona CA, Behrens A. ATM signalling and cancer. Oncogene 2013; 33:3351-60; PMID:23851492; http://dx.doi.org/10.1038/onc.2013.275
- Herrero AB, San-Miguel JF, Gutierrez NC. Deregulation of DNA double-strand break repair in multiple myeloma: implications for genome stability. PLOS One 2015; 10:e0121581; PMID:25790254; http://dx.doi.org/10.1371/journal.pone.0121581
- Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 2011; 474:230-4; PMID:21654808; http://dx.doi.org/10.1038/nature10155
- Sola B, Troussard X. Relevance of cyclin D1 level in the pathogenesis of multiple myeloma. Blood 2003; 102:4245-6; PMID:14623774; http://dx.doi.org/10.1182/blood-2003-07-2486
- Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, Morgan G, Van Ness B, Chesi M, Minvielle S, et al. International myeloma working group molecular classification of multiple myeloma: spotlight review. Leukemia 2009; 23:2210-21; PMID:19798094; http://dx.doi.org/10.1038/leu.2009.174
- Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, Thuret JY, Andrews S, Fraser P, Reik W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep 2015; 10:471-83; PMID:25640177; http://dx.doi.org/10.1016/j.celrep.2014.12.055
- Corpet A, Stucki M. Chromatin maintenance and dynamics in senescence: a spotlight on SAHF formation and the epigenome of senescent cells. Chromosoma 2014; 123:423-36; PMID:24861957; http://dx.doi.org/10.1007/s00412-014-0469-6
- Wang Y, Liang Y, Vanhoutte PM. SIRT1 and AMPK in regulating mammalian senescence: a critical review and a working model. FEBS Lett 2011; 585:986-94; PMID:21130086; http://dx.doi.org/10.1016/j.febslet.2010.11.047
- Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep 2014; 15:1139-53; PMID:25312810; http://dx.doi.org/10.15252/embr.201439245
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013; 123:966-72; PMID:23454759; http://dx.doi.org/10.1172/JCI64098
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Dual mTORC1/C2 inhibitors suppress cellular geroconversion (a senescence program). Oncotarget 2015; 6:23238-48; PMID:26177051; http://dx.doi.org/10.18632/oncotarget.4836
- Maiso P, Liu Y, Morgan B, Azab AK, Ren P, Martin MB, Zhang Y, Liu Y, Sacco A, Ngo H, et al. Defining the role of TORC1/2 in multiple myeloma. Blood 2011; 118:6860-70; PMID:22045983; http://dx.doi.org/10.1182/blood-2011-03-342394
- Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer 2015; 15:397-408; PMID:26105537; http://dx.doi.org/10.1038/nrc3960
- Gallego ME, White CI. RAD50 function is essential for telomere maintenance in Arabidopsis. Proc Natl Acad Sci USA 2011; 98:1711-6; http://dx.doi.org/10.1073/pnas.98.4.1711
- Le S, Moore JK, Haber JE, Greider CW. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 1999; 152:143-52; PMID:10224249
- Shiratsuchi M, Muta K, Abe Y, Motomura S, Taguchi F, Takatsuki H, Uike N, Umemura T, Nawata H, Nishimura J. Clinical significance of telomerase activity in multiple myeloma. Cancer 2002; 94:2232-8; PMID:12001122; http://dx.doi.org/10.1002/cncr.10472
- Halicka HD, Zhao H, Li J, Hsieh TC, Wu JM, Darzynkiewicz Z. Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling. Aging 2012; 4:952-65; PMID:23363784; http://dx.doi.org/10.18632/aging.100521
- Laurent C, Leduc A, Pottier I, Prévost V, Sichel F, Lefaix JL. Dramatic increase in oxidative stress in carbon-irradiated normal human skin fibroblasts. PLOS One 2013; 8:e85158; PMID:24376870; http://dx.doi.org/10.1371/journal.pone.0085158
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 2008; 3:1101-8; PMID:18546601; http://dx.doi.org/10.1038/nprot.2008.73