
ABSTRACT
Lineage specification of both mouse and human pluripotent stem cells (PSCs) is accompanied by spatial consolidation of chromosome domains and temporal consolidation of their replication timing. Replication timing and chromatin organization are both established during G1 phase at the timing decision point (TDP). Here, we have developed live cell imaging tools to track spatio-temporal replication domain consolidation during differentiation. First, we demonstrate that the fluorescence ubiquitination cell cycle indicator (Fucci) system is incapable of demarcating G1/S or G2/M cell cycle transitions. Instead, we employ a combination of fluorescent PCNA to monitor S phase progression, cytokinesis to demarcate mitosis, and fluorescent nucleotides to label early and late replication foci and track their 3D organization into sub-nuclear chromatin compartments throughout all cell cycle transitions. We find that, as human PSCs differentiate, the length of S phase devoted to replication of spatially clustered replication foci increases, coincident with global compartmentalization of domains into temporally clustered blocks of chromatin. Importantly, re-localization and anchorage of domains was completed prior to the onset of S phase, even in the context of an abbreviated PSC G1 phase. This approach can also be employed to investigate cell fate transitions in single PSCs, which could be seen to differentiate preferentially from G1 phase. Together, our results establish real-time, live-cell imaging methods for tracking cell cycle transitions during human PSC differentiation that can be applied to study chromosome domain consolidation and other aspects of lineage specification.
Introduction
Eukaryotic DNA replication follows a defined spatial-temporal sequence that is achieved by the nearly synchronous firing of clusters of origins along 400–800 kb replication domains (RDs). Genome-wide studies have shown that this “replication timing program” is cell type specific and highly conserved between related species.Citation1-3 RDs that replicate early correlate with transcriptional activity, suggesting that replication timing reflects other chromosome functions.Citation4-6 During development many RDs undergo changes in replication timing that are accompanied by changes in subnuclear position and transcriptional competence.Citation2 In each cell cycle, replication timing is re-established coincident with the anchorage of chromosome domains at a discrete time during G1 termed the timing decision point (TDP),Citation7,8 demonstrating an intimate relationship between sub-nuclear position and replication timing.
Cytogenetically, DNA replication can be observed to occur at discrete punctate sites, called replication foci, the spatial distribution of which changes characteristically during the course of S phase.Citation9 During early S phase replication foci are enriched in the interior of the nucleus, whereas replication foci appearing in late S phase are enriched along the periphery of the nucleus and nucleoli, and other heterochromatic regions.Citation9 Individual replication foci labeled with nucleotide analogs and followed over numerous generations do not diminish in size or intensity, indicating that the DNA replicated within individual foci remains stably associated through many cell cycles.Citation10-13 Thus, replication foci are stable chromosome units that are likely the equivalent of RDs identified by genomics methods, although this has not been directly demonstrated.
3D maps of chromatin interactions (Hi-C) and lamina-associating domain (LAD) mapping have revealed a strong correlation between sub-nuclear position, chromatin interaction compartments and replication timing, providing molecular confirmation of this spatio-temporal organization of chromatin.Citation1,14 In addition, Hi-C mapping has uncovered structures within chromatin compartments known as topologically associated domains (TADs).Citation15 We have recently shown that TADs share chromosomal boundaries with RDs, suggesting that TADs, RDs and replication foci are reflections of the same developmentally stable, large-scale chromatin structures.Citation16 Altogether, these observations suggest a model (“replication domain model”) in which RDs and TADs are equivalent units of chromosome structures, and the 3D folding of TAD/RDs creates compartments in which TAD/RDs in close proximity replicate at similar times and can be visualized as punctate replication foci.
Both human and mouse embryonic stem cells (hPSCs and mESCs) exhibit a unique RD organization in which a higher percentage of adjacent RDs replicate discordantly.Citation5,17 Upon differentiation RDs rapidly consolidate, both temporally to form larger coordinately replicated constant timing regions (CTRs), and spatially to form larger blocks of dense chromatin.Citation5,17 This consolidation was recently confirmed by Hi-C mapping,Citation18 but its biological significance remains unknown. We have previously demonstrated that replication timing is established during G1 coincident with the anchorage of chromosome positions inside of the nucleus, and the formation of TADs and sub-nuclear compartments.Citation7,19 However, hPSCs and mESCs are known to have an abbreviated G1 phase, which is lengthened upon differentiation.Citation20-23 Within this context, it is conceivable that pluripotent cells initiate replication prior to the complete re-establishment of sub-nuclear compartments, while the lengthening of G1 phase during differentiation could provide time for adjacent RDs to assemble into more consolidated compartments.Citation24 Global reorganization of chromosomal domains, consolidation and accumulation of heterochromatin may contribute to the stable silencing of genes that are no longer required or are detrimental to lineage specification.Citation25
Although the existence of spatio-temporal patterns of replication foci has been documented in mESCs,Citation26 the dynamic organization of these patterns during differentiation has not been addressed. In this study we develop a live cell imaging system to track the reorganization of RDs in single PSCs as they undergo cell-fate transitions. We find significant changes in the spatio-temporal patterns of replication foci during differentiation consistent with RD consolidation. However, these changes occurred without any detectable lengthening of G1 phase. We employ our live cell imaging system to show that anchoring of domains is accomplished within the abbreviated G1, putting to rest the hypothesis that hPSCs' unique chromosome domain structure is a consequence of replication initiating prior to the complete reorganization of RDs during G1. Finally, we exploited this system to study the cell cycle regulation of cell fate choice, confirming with single cell analyses that hPSCs preferentially respond to induction factors during G1.
Results
The Fucci system does not demarcate the G1 to S phase transition
We aimed to examine dynamics of spatio-temporal organization of replication foci in real time, which required a means to identify S-phase cells. The Fucci cell cycle indicator systemCitation27 reports cell-cycle phases using fluorescent proteins, Kusabira-Orange 2 (KO2) and Azami-Green-1 (Az1), fused to Skp, Cullin, F-box containing complex (SCF) and anaphase-promoting complex (APC) targeted degrons derived from Cdt1 and Geminin, respectively. Cell cycle phases are then reported based on the accumulation vs. targeted destruction of these 2 cell-cycle-regulated proteins. The Fucci system is illustrated schematically in , and has recently been adapted for hPSCs ().Citation28 We assessed the ability of this system to faithfully report cell-cycle transitions in hPSCs by directly comparing the expression of Fucci reporters to the presence or absence of a synthetic nucleotide analog, EdU, as well as the detergent-resistant chromatin association of the mini-chromosome maintenance (MCM) helicase subunit Mcm5 (). MCMs are loaded onto chromatin during telophase, tightly bound to chromatin throughout G1 phase, and removed from active replication forks throughout the course of S phase,Citation29 while G2 phase cells completely lack detergent-resistant Mcm. H9 Fucci expressing cells were briefly pulse labeled with EdU and expression of Fucci reporters was photographed by live cell fluorescent microscopy. Thereafter, soluble Fucci proteins were extracted by a brief triton wash and the same cells were stained for EdU and MCM and re-imaged. Results revealed that ∼10% of KO2+ cells had initiated DNA replication (EdU+) with early S phase patterns (). Thus, degradation of the KO2 reporter occurs distinctly after entry into S phase.
Figure 1. Single-cell evaluation of Fucci in hPSCs. (A) Diagram of Fucci reporters' anticipated expression patterns during the cell cycle. (B) Diagram of adapted Fucci reporters driven by the PSC-expressed CAG promoter and linked with selectable markers through an internal ribosome entry site (IRES). (C) Fluorescent microscopy images directly comparing Fucci reporters (pre-extract, top panels) to cell cycle specific markers MCM5 and EdU (post-extract, lower panels) within the same cells. Fucci expressing hPSCs were pulse labeled with EdU prior to imaging. One KO2+ cell is EdU+ indicating this cell initiated replication before degradation of KO2. (D) Table comparing Fucci expressing hPSCs (Fucci expression reported on rows, DN = double negative, DP = double positive) to cell cycle position based on the presence/absence of EdU and extraction-resistant MCM5 (columns).
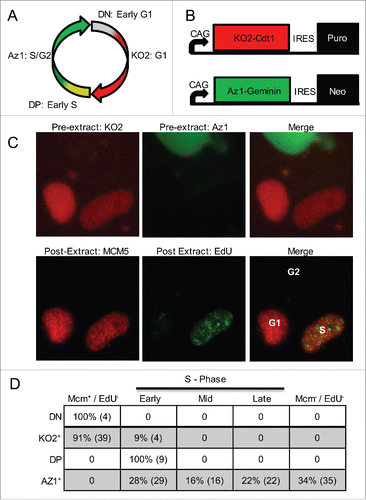
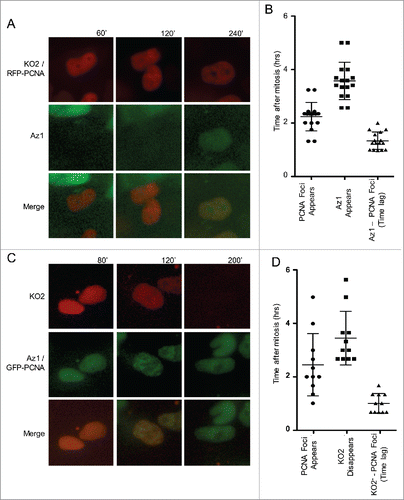
To confirm this result we transfected Fucci expressing cells with a fluorescent tagged replication fork protein, PCNA, which forms prominent replication foci upon entry into S phase, and conducted live-cell imaging experiments. Our results reveal that PCNA foci appear approximately 1 hr before the accumulation of the Az1-tagged APC-degron for geminin () and the targeted destruction of the SCF-degron derived from Cdt1 (), confirming that, in hPSCs, entry into S phase precedes the transition in Fucci reporters. Interestingly, these results are consistent with an earlier report that geminin does not accumulate until several hours after the onset of S phase in Chinese Hamster fibroblasts,Citation30 suggesting that geminin is not necessary to prevent re-replication during early S phase. Together, our results demonstrate that Fucci is not able to identify the G1/S transition in hPSCs. Since Fucci is also unable to identify the S/G2 or G2/M transitions, we conclude it is not useful to measure cell cycle phase lengths.
Figure 2. The Fucci system does not accurately designate the G1 to S phase transition. (A) Panels taken from a live-cell-imaging video of Fucci expressing hPSCs transiently transfected with RFP-PCNA. Top panels correspond to KO2 & RFP-PCNA, middle panels correspond to Az1. PCNA foci appear prior to the accumulation of Az. (B) Quantification of Time (in hours) after mitosis that PCNA foci and Az1 are detected in live cell imaging videos. PCNA foci appear ∼1 hr prior to the detection of Az1. (C) Panels from a live-cell-imaging video of Fucci expressing hPSCs transiently transfected with GFP-PCNA. Top panels correspond to KO2, middle panels are GFP-PCNA & Az1. PCNA foci appear prior to the disappearance of KO2-Cdt1. (D) Quantification of Time (in hours) after mitosis that PCNA foci are detected and KO2-Cdt1 signal disappears in live-cell imaging videos. PCNA foci appear ∼1 hr prior to the disappearance of KO2-Cdt1.
An improved imaging system for live cell imaging studies of replication in hPSCs
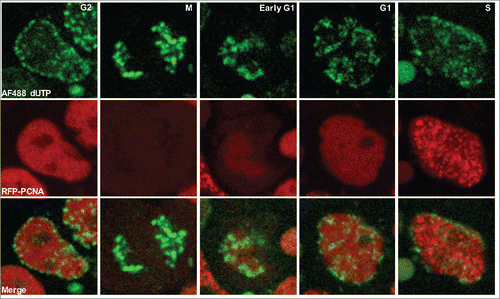
PCNA has been used to image replication foci and track their spatio-temporal changes during S phase in living cells.Citation31 We reasoned that the use of fluorescently tagged PCNA, coupled with visible changes in cell morphology during mitosis, would be sufficient to track all the transitions in the phases of the cell cycle in hPSCs, in addition to tracking the spatio-temporal changes in replication foci. We transfected H9 hPSCs transiently with RFP-PCNA and subjected the cells to long-term, live-cell imaging. To track multiple cells simultaneously, and to reduce phototoxicity, long term imaging was performed at low magnification using an Olympus VivaView incubator microscope. When conducting live cell imaging experiments we selected cells with moderate to low levels of RFP expression to avoid potential toxic effects of PCNA over-expression. In addition, we confirmed that expression of PCNA at these observed levels did not impair the cell cycle by comparing the doubling times of hPSCs transfected with RFP-PCNA, to those expressing RFP-H2B or untagged RFP (Fig. S1). Cell cycle lengths in all cases were similar and consistent with previously published methodsCitation21,23,32 reporting an average doubling time of ∼18 hrs, indicating that the ectopic expression of RFP-PCNA does not impact overall cell cycle length. As the panels in show, we were able to identify cell cycle phases by monitoring PCNA foci relative to cell divisions. Furthermore, even at low magnification, we were able to distinguish the changes in spatio-temporal replication foci patterns during S phase, which are consistent with previous reports and our higher-resolution images ( and Fig. S2).Citation9,26,31 In the first replication foci pattern, Pattern I, sites of replication are distributed throughout the interior, euchromatic, nucleoplasm, with little proximity to the nucleolus or nuclear periphery. After ∼5hrs, Pattern II emerges in which sites of replication become apparent around the periphery of nucleus and nucleoli, and the number of foci in the interior of the nucleus is reduced. In Pattern III, replication foci are further depleted in the interior while regions of replication around the periphery of the nucleus and nucleoli become more prominent. By Pattern IV, sites of replication become larger in size and fewer in number and are distributed within the nuclear interior. Finally, at the end of S phase, the last pattern of replication emerges, Pattern 5, in which there are still fewer and larger sites of replication visible.
Figure 3. An improved imaging system for live cell imaging studies of replication in hPSCs. (A) Panels from a live-cell-imaging video of RFP-PCNA expressing hPSCs with deduced cell cycle position indicated. Mitosis can easily be detected by drastic morphological changes to nuclei. G1 phase cells lack PCNA foci but PCNA foci are readily detectable upon entry into S phase. The 5 distinct spatial temporal foci patterns observed in hPSCs are indicated in the panels. G2 phase cells can be detected either by the disappearance of replication foci, or based on subsequent entry into mitosis. (B) Individual cell cycle phase lengths recorded from live-cell imaging videos of RFP-PCNA expressing H9 hPSCs. (C) Table of the cell cycle parameters for cell lines examined. Numbers indicate hours for each phase.
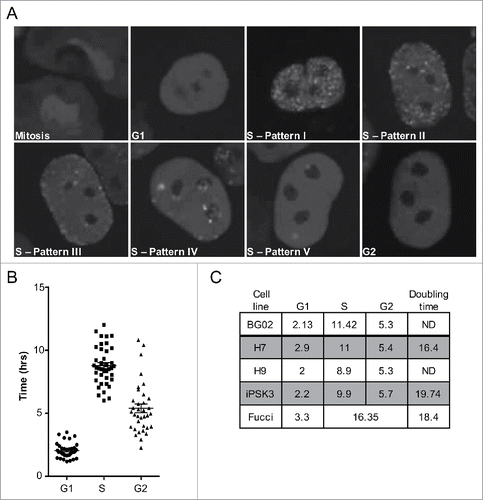
The appearance and disappearance of replication foci relative to cell division by definition demarcates the G1/S & S/G2 boundaries, respectively. Thus, by transiently transfecting cells with our RFP-PCNA construct and performing live-cell imaging we were able to determine the exact lengths of G1, S, and G2 phases in individual hPSCs. We show that H9 hPSCs have an abbreviated G1 of ∼2 hrs, S phase ∼9 hrs, and G2 phase is ∼5 hrs (). We extended this analysis to include multiple hESC cell lines as well as an induced pluripotent stem cell (iPSC) line and show that this unique profile is highly conserved among hPSCs (). These results are corroborated by fluorescence activated cell sorting (FACS) analysis of H9 hPSCs stained for DNA content, which confirm a smaller percentage of cells are present in G1 than S or G2 phase (Data not shown). Importantly, our results are the first direct measurements of G1 length in human pluripotent stem cells.
Spatio-temporal re-organization of replication foci accompanies RD consolidation
Both human and mESCs share a unique chromatin organization characterized by more discordantly replicating RDs,Citation5,17 and less clustered TADsCitation18 that consolidate upon differentiation. Since spatio-temporal patterns of DNA replication are a reflection of the 3D organization of chromatin, we reasoned that domain consolidation during differentiation might be detectable as changes in the amount of the genome organized within the various spatio-temporal replication patterns. For instance, since pattern 1 is characterized by a more dispersed distribution of replication foci, domain consolidation would be predicted to result in a lower fraction of the genome organized within replication pattern 1 in differentiated cells compared to PSCs. We chose the definitive endoderm (DE) differentiation system as our model because of the rapid and robust differentiation scheme available.Citation33 hPSCs stimulated by high concentrations of Activin A reach DE in approximately 48 hrs, after passing through a mesendoderm intermediate. The differentiation protocol is outlined in . To monitor for differentiation in single cells we employed an endoderm reporter cell line, H9 Sox17-mCherry, in which the Sox17 promoter drives mCherry expression.Citation34 Several lines of evidence establish that under these conditions Sox17 is a reliable marker for definitive endoderm differentiation.Citation35-37 In our hands we detect ∼70% mCherry+ cells following 48 hrs of stimulation ().
Figure 4. Sox17-mCherry reporter line exhibits consolidation of RDs upon differentiation. (A) Table of differentiation scheme and diagram showing established cell type markers. (B) Fluorescent microscopy images of H9 Sox17-mCherry cells undifferentiated (top panels) and after 48 hrs of differentiation (lower panels) taken at 20x magnification. (C) Schematic of genome-wide replication timing (Repli-seq) protocol. (D) Replication timing profiles of chromosome 8 for ESCs and DE. Regions of consolidation are outlined by boxes.
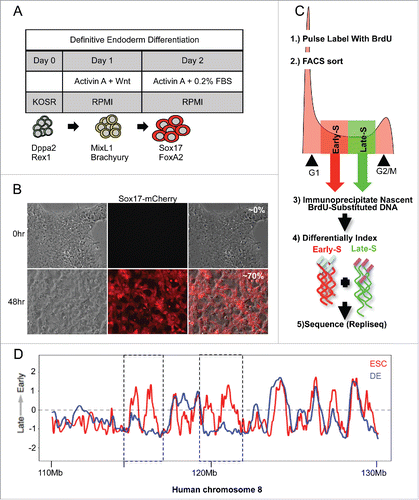
Next, before investigating the association of G1 length and RD reorganization in single cells, we confirmed the consolidation of domains in our reporter cell line under our differentiation conditions by genome-wide replication timing analysis (). Briefly, populations of unstimulated cells and cells stimulated for 48 hrs (DE) were pulse labeled with 5′-bromo-2′-deoxyuridine (BrdU), retroactively synchronized into early and late S phase factions by flow cytometry, then BrdU incorporated DNA from early and late S phase fractions was immunoprecipitated and sequenced. Sequences were mapped back to the genome in 6kb bins and plotted as a ratio of early to late replicating DNA. We detected changes in genome-wide replication timing that are consistent with differentiation to DECitation5 (Fig. S3) and confirmed consolidation of RDs in our reporter cell line. shows an exemplary region of chromosome 8 in which smaller domains that replicate at different times in PSCs merge to form larger coordinately replicated domains upon differentiation.
To test the hypothesis that domain consolidation can be visualized as a reduction in the duration of replication pattern 1, we differentiated H9 Sox17-mCherry cells harboring a GFP-PCNA reporter and measured the percentage of S phase cells displaying each replication pattern. Results revealed that, upon differentiation, there is a significant decrease in the percent of S phase dedicated to replicating pattern 1 and a corresponding increase in the duration of later patterns (). These results demonstrate that consolidation of domains during ESCs differentiation observed by genomics methods coincides with a spatial consolidation of replication foci observed cytogenetically by dynamically tracking spatio-temporal patterns.
Figure 5. Spatio-temporal re-organization of replication foci accompanies differentiation without G1 phase lengthening. The % of S phase spent replicating each pattern for GFP-PCNA expressing H9 Sox17-mCherry cells using live-cell-imaging videos (Exemplary patterns shown in ). Undifferentiated hPSCs are in black, differentiated (mCherry+) cells are in red. G1 lengths of cells observed are shown in the inset of figure. *p value < 0.05.
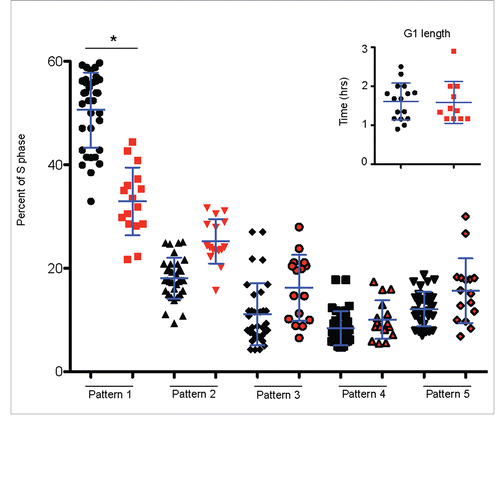
Both human and mouse PSCs share an abbreviated G1 that has been claimed to lengthen during differentiation.Citation21-23,38 As we discussed in the introduction, in principle, a reasonable explanation for domain consolidation is that the longer G1 phase in differentiated cells could permit more time for domains to consolidate before S phase begins.Citation24 However, previous studies measuring G1 phase length of ESCs employed either the Fucci systemCitation21 to measure G1 length, which we show in does not detect the G1 to S phase transition, or prolonged drug-induced cell cycle arrest,Citation22 which perturbs cell cycle structure. To investigate whether domain consolidation accompanies a lengthened G1 phase, we measured G1 length in the same cells used to track replication foci patterns. Results indicated that G1 length did not increase during the course of 48 hours of differentiation within these individual cells that induced Sox17-RFP (, inset). Our results are not necessarily inconsistent with G1 phase lengthening during differentiation observed by others. It is possible that differentiation for a longer period of time may lengthen G1. We also would not have scored cells that had exited the view field, and cells with longer G1 phases would have had more time to exit the view field. Regardless, our results demonstrate that, changes in the spatio-temporal replication patterns occur without an accompanying increase in G1 length.
hPSC RD re-organization following mitosis is complete prior to DNA replication
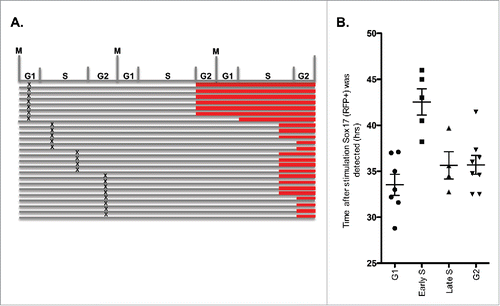
The results presented above show domain consolidation occurs despite the maintenance of a short G1. Thus, either 3D positioning does not need to be completed before S phase begins, or RDs re-position within the abbreviated G1 phase time. In fibroblasts, re-positioning takes 2–3 hours after mitosis, which is close to the length of an ESC G1 phase.Citation7,19 To distinguish these possibilities, we labeled RDs by pulse-labeling RFP-PCNA expressing hPSCs with fluorescently labeled nucleotides. hPSCs in mid-S phase at the time of labeling exhibited a characteristic mid-S pattern of replication foci that are relatively immobile throughout interphase. This pattern was easily distinguished from the random chromatin 3D organization and rapid movement of replication foci seen immediately after nuclear membrane re-assembly.Citation39 We therefore tracked cells with labeled mid-S replication foci through mitosis and into the next G1 phase using live-cell microscopy, and related the spatial organization and movement of those foci to the appearance of PCNA foci in the same cells, indicative of the onset of S phase. This experiment revealed that, despite the abbreviated G1 phase, chromosome positions and restricted motion of foci were completed prior to the appearance of PCNA foci (). Interestingly, when we repeated this experiment using Fucci expressing hPSCs, we found that the KO2-tagged Cdt1-degron was stabilized prior to the re-establishment of chromosomal positions (Fig. S4), indicating that double-negative Fucci cells can be employed to study cell cycle events that occur prior to the re-establishment of chromosome organization and replication timing at the TDP.
Figure 6. hPSC RD re-organization following mitosis is complete prior to DNA replication. Panels taken from a live cell imaging video of RFP-PCNA expressing hPSCs with fluorescently labeled RDs. RDs are labeled by briefly pulse-labeling hPSCs with green fluorescent conjugated nucleotides, which are rapidly incorporated into replicating DNA. Using live-cell-imaging, a cell in Mid-S phase at the time of pulse-labeling was identified based on its Mid-S phase foci pattern (middle panels) and tracked through mitosis until the subsequent S phase, indicated by the appearance of RFP-PCNA foci (top panels). Similar results were obtained in 20 cells.
Sox17 is preferentially induced from G1 phase
Recent evidence indicates that hPSCs are more responsive to induction factors during G1.Citation40-43 However, the intrinsic cell to cell variation in responsiveness has not been addressed. Our live cell imaging approach provided an opportunity to investigate the relationship between cell cycle position and differentiation in single cells, in real time. We tracked the appearance of Sox17-mCherry in cells stimulated to differentiate and traced each cell back to identify the cell-cycle phase of each individual cell when differentiation factors were added (time = 0). Our results show that hPSCs in G1 phase at the time of induction induced Sox17 in a cell cycle earlier () and more rapidly (), than cells in S or G2 phase. Cells in early S took the longest to induce Sox17, suggesting they must come around a full cell cycle to the next G1 before they can elicit a response to induction factors. These results demonstrate the utility of our novel system to monitor cell cycle phase transitions during cell fate commitment.
Figure 7. Sox17 is preferentially induced from G1 phase. (A) Diagram of cell cycle phase induction of Sox17-mCherry in hPSCs. Observations were recorded from live-cell-imaging videos of Sox17-mCherry, GFP-PCNA expressing hPSCs. mCherry+ cells were traced back to time = 0, the stage of cell cycle that the cell was positioned at time of stimulation was deduced from GFP-PCNA as described in . The cell cycle phase that the cell was stimulated is indicated by “X.” Point at which cell became mCherry+ indicated by red bar. (B) Quantification of when cells were turned mCherry+ (Y axis, time in hours) relative to phase of cell cycle from which cells were positioned at time of induction (indicated on X axis).
Discussion
We have developed a system to track RDs during the cell cycle in human PSCs in real time and we have investigated whether chromosome domain consolidation during human PSC differentiation is related to G1 lengthening. Our results represent the first characterization of replication patterns in living cells during differentiation. We show that the spatial distribution and temporal organization of DNA replication foci changes in a manner that is consistent with the consolidation of RDs detected by genome-wide RTCitation5,17 and Hi-C analysis.Citation18 Furthermore, we establish that domain consolidation can occur without an accompanying increase in G1 phase length. This finding puts to rest a long-standing hypothesis that the more discordant replication and compartmentalization of RDs observed in PSCs is a consequence of replication origins firing prior to the complete spatial re-organization of RDs.Citation24
Biological significance of domain consolidation
We conclude that G1 lengthening does not account for domain consolidation; although, the significance of this unique domain structure is still unclear. The conservation of more discordantly replicated and spatially separated RDs in both human and mouse PSCs suggests consolidation serves some role in lineage committment.Citation5,17 Discordantly replicating RDs may reflect a specialized feature of PSC chromosome structure in which domains have flexibility to organize into different types of more restricted spatial-temporal patterns during lineage specification. Spatio-temporal consolidation could provide a scaffold to assemble more stable epigenetic states that define cell lineages. While their exact role is still unclear, RDs reflect meaningful aspects of chromosome biology and the study of RD dynamics is likely to reveal an important molecular handle for executing proper differentiation programs. Our results demonstrate that this consolidation is not a passive result of the time permitted for chromatin to re-organize but is a feature of the pluripotent cell interphase chromatin architecture.
Replication foci and replication domains
The preponderance of evidence suggests that replication foci correspond to the replication domains seen by genome-wide replication timing analyses, which are equivalent to TADs.Citation16 Once labeled, replication foci remain visible as stable units for many generations.Citation10,12,13 DNA fiber studies reveal that RDs replicate by the synchronized firing of clusters of origins across hundreds of kilobases.Citation44-47 Moreover, the estimated amount of DNA per replication focusCitation46 is consistent with the sizes of RDs, and replication foci appear quite uniform in size, similar to RDs, which range from 400–800 kb.Citation1,17 The sizes of coordinately replicating segments of chromosomes in any give cell type on the other hand are far less uniform in size, ranging from 400 kb to several Mb, due to the coordinate replication of adjacent RDs. In fact, studies of PCNA in living cells have shown that, frequently, upon the completion of replication at one focal site, replication initiates rapidly within an adjacent site, which would give rise to a large segment of coordinated DNA replication (or a large CTR) in the ensemble genome-wide data.Citation48 Consolidation during differentiation results in an increase in the number of adjacent RDs that are coordinately replicating and spatially consolidated.Citation5,17 Our results here show that, during differentiation, replication foci spatially consolidate to reduce the amount of time cells spend replicating the more disperse pattern I foci, coincident with domain consolidation measured by other methods. Altogether, this gives rise to a model in which replication foci are the cytogenetic equivalent of replication domains seen by genome-wide replication timing maps, and TADs seen in Hi-C data, and clusters of adjacent replication foci/TADs/RDs replicate at very similar times, giving the appearance of larger CTRs.Citation49
Differentiation from G1 phase
We have employed our live single cell imaging system to show that PSCs induce definitive endoderm marker Sox17 more rapidly when stimulated prior to the onset of DNA replication. This result is consistent with ensemble methods that have reported that PSCs' capacity to differentiate is significantly reduced as cells exit G1 phase.Citation40-42 The prevailing view is that G1 represents a critical opportunity for cells to elicit a response to differentiation signals, and once cells pass this window of opportunity, cells are refractory to such factors until the next cell cycle. Moreover, recent evidence shows that undergoing DNA replication is a requirement for successful cellular reprogramming and gene repositioning.Citation50-52 It is tempting to speculate that passage through S phase is required to remodel chromatin as it is repackaged at the replication fork. Indeed, different types of chromatin replicate at different times during S phase, thus, a switch in replication timing could facilitate a domain-wide remodeling of chromatin. Additionally, the replication machinery has been shown to interact with chromatin remodelersCitation53,54 and components of replication machinery have been identified as important factors in gene positioning.Citation51 Thus it is possible that cells experiencing differentiation signals during G1 elicit a response via changes to the replication-timing program. This model would explain correlations observed between RT, chromatin organization, and transcription. It also raises the interesting question as to the order in which these events change during a single cell type transition. Addressing this question will bring us closer to understanding how these events are casually linked and provide a foundation for future studies of mechanism.
Materials and methods
Cell culture
Cell lines H7, H9, BG01, BG02, and iPSK3 were cultured under feeder-free conditions on Geltrex (Thermo Fisher, A14133) coated dishes, and maintained in StemPro (Thermo Fisher, A100701) culture media per manufacture's specifications. Cell passaging was achieved by brief treatment (∼6–8 min) with Accutase (Thermo Fisher, A1110501). After detachment, cells were gently collected, centrifuged for 5 min at 200 g, and re-plated on freshly coated Geltrex dishes. The H9 derived Sox17-mCherry reporter cell line was provided by our collaborators Edouard Stanley and Andrew Elefanty and maintained in DMEM F12 (Invitrogen, 11320–033), 20% Knockout Serum Replacement (Invitrogen, 10828–028), 2 mM Glutamax (Invitrogen, 35050–061), 100uM Nonessential amino acids (Invitrogen, 11140–050), and 10ng/ml bFGF (Thermo Fisher, PHG0261).
Definitive endoderm differentiation
Detailed differentiation conditions may be found at Schultz et al., 2010. Briefly, cultures at ∼70% confluency were washed 2x with cold PBS and stimulated to differentiation by the addition of RPMI (Invitrogen), 50ng/ml Wnt (CellGS, GFM77), 100ng/ml Activin A (CellGS, GFH6). 24hrs after stimulation media was replaced with RPMI containing 0.2% FBS, and 100 ng/ml Activin A.
Generation of cell cycle reporter lines
The FUCCI reporter plasmids (described in ) were generously provided by Amar Sigh (Steve Dalton's lab, UGA). Plasmids were transfected into hPSCs using FugeneHD (Promega, E2311) following manufactures specifications. Four days following transfection, Fucci expression hPSCs were selected based on resistance to antibiotic drugs Neomycin and Puromycin. Drug-resistant colonies were pooled. RFP-PCNA and GFP-PCNA plasmids were provided by Michael Davidson (FSU). Plasmids were transfected into hPSCs using FugeneHD following manufactures guidelines.
Immunofluorescence
To compare Fucci with established cell cycle markers EdU and MCM5, H9 Fucci expressing hPSCs were pulse labeled with EdU (10 uM) for 20 min and subsequently imaged using an image restoration microscope system (DeltaVision; Applied Precision) attached to a fluorescence microscope (Olympus, IX-71) equipped with a Plan Apo1.40 NA oil objective lens (Olympus). Stage positions where images were captured were recorded. Soluble Fucci reporters were then extracted by 0.5% Trition X-100 diluted in ice-cold CSK buffer supplemented with protease inhibitors (1:50, Cocktail III, VWR, 80053–852). Following extraction, cells were fixed by incubating cells in 4% paraformaldehyde for 15min on ice. Cells were washed 3x with cold PBS and incubated in blocking buffer containing: 10% normal goat serum, 0.2% BSA, diluted in cold PBS for 1hr. DNA-incorporated EdU was then labeled by Click-it Alexa Fluor 488 (Thermo Fisher, C10337). MCM5 was labeled by incubating cells for 1hr with MCM5 primary antibody (1:100, Santa Cruz) diluted in blocking buffer. Cells were washed 3x with cold PBS and incubated for 1hr in blocking buffer containing secondary antibody Alexa Fluor 594. Cells were washed and stained with DAPI (2 ug/ml). Stage positions were re-visited using Olympus Deltavision software and cells were re-imaged.
Long-term, live-cell imaging of cell cycle and RDs
For imaging experiments in which cell cycle parameters and replication foci patterns were recorded, cells were plated onto Geltrex-coated, glass-bottom dishes (MatTek). Live cell imaging was performed with a fluorescent incubator microscope (Olympus VivaView FL) equipped with a 40x objective and a highly sensitive EM-CCD camera (Hamamatsu, ImagEM). Time-lapse images were collected every 12–20 min for 48 hrs at minimal exposure times to avoid phototoxicity. Image analysis was performed using the Olympus VivaView software package, Metamorph. S-phase replication foci patterns were visually identified.
Genome-wide replication timing analysis
Genome-wide RT profiles were constructed as described previously.Citation17 Array hybridization was replaced by next generation sequencing (NGS) (Repli-seq). We have previously shown Repli-seq produces indistinguishable results from array hybridization.Citation16 RT datasets were normalized using limma package in R (R Core Team 2015) and rescaled to equivalent ranges by quintile normalization.
Tracking re-organization of RDs after mitosis
To track the re-establishment of RDs following mitosis, cells were labeled with Alexa Fluor 488 dUTP (Thermo Fisher, C11397). For this purpose, cells were seeded onto 35 mm glass bottom dishes (MatTek) and transfection was performed at 70% confluency. For each dish, 3 μl Fugene6 (Promega, E2691) was mixed with 1 μl dUTP analog (10 minutes; 0°C) and 17 μl PBS (further 5 minutes; 0°C) before applying to cells (10 minutes; 0°C). Dishes were rinsed with PBS and growth media was returned. Cells were imaged using either the Olympus VivaView, or Andor Revolution Spinning disk equipped with 60x oil objective and Andor Clara camera for high resolution image capture.
Abbreviations
| APC | = | Anaphase Promoting Complex |
| Az1 | = | Azami-Green 1 |
| BrdU | = | 5′-bromo-2′-deoxyuridine |
| CTR | = | Constant timing region |
| DE | = | Definitive Endoderm |
| Fucci | = | Fluorescence ubiquitination cell cycle indicator |
| GFP | = | Green Fluorescent Protein |
| hESC | = | Human embryonic stem cell |
| KO2 | = | Kusabira Orange 2 |
| LADs | = | Latin associated domain |
| MCM | = | Mini-chromosome maintenance |
| mESC | = | Mouse embryonic stem cell |
| PSCs | = | Pluripotent stem cells |
| PCNA | = | Proliferating Cell Nuclear Antigen |
| RFP | = | Red Fluorescent Protein |
| RD | = | Replication domain |
| SCF | = | Skp, Cullin, F-box containing complex |
| TAD | = | Topologically associated domain |
| TDP | = | Timing decision point |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Notes on contributors
Manuscript writing: KAW; Conception and design: KAW and DMG; Reporter cell line construction: AGE and EGS; data analysis and interpretation: KAW and DMG; Manuscript editing: KAW, AGE, EGS, DMG
Supplementary files
Download Zip (519 KB)Acknowledgements
We thank H. Bass and V. Dileep for helpful discussions.
Funding
This work was funded by NIH grant GM085354 to DMG.
Related Research Data
References
- Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 2010; 20:761-70; PMID:20430782; http://dx.doi.org/10.1101/gr.099655.109
- Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, Fussner E, Bazett-Jones DP, Plath K, Dalton S, et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res 2010; 20:155-69; PMID:19952138; http://dx.doi.org/10.1101/gr.099796.109
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A 2010; 107:139-44; PMID:19966280; http://dx.doi.org/10.1073/pnas.0912402107
- Hiratani I, Takebayashi S, Lu J, Gilbert DM. Replication timing and transcriptional control: beyond cause and effect–part II. Curr Opin Genet Dev 2009; 19:142-9; PMID:19345088; http://dx.doi.org/10.1016/j.gde.2009.02.002
- Rivera-Mulia JC, Buckley Q, Sasaki T, Zimmerman J, Didier RA, Nazor K, Loring JF, Lian Z, Weissman S, Robins AJ, et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res 2015; 25:1091-103; PMID:26055160; http://dx.doi.org/10.1101/gr.187989.114
- Lubelsky Y, Prinz JA, DeNapoli L, Li Y, Belsky JA, MacAlpine DM. DNA replication and transcription programs respond to the same chromatin cues. Genome Res 2014; 24:1102-14; PMID:24985913; http://dx.doi.org/10.1101/gr.160010.113
- Dimitrova DS, Gilbert DM. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell 1999; 4:983-93; PMID:10635323; http://dx.doi.org/10.1016/S1097-2765(00)80227-0
- Pope BD, Hiratani I, Gilbert DM. Domain-wide regulation of DNA replication timing during mammalian development. Chromosome Res 2010; 18:127-36; PMID:20013151; http://dx.doi.org/10.1007/s10577-009-9100-8
- O'Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol 1992; 116:1095-110; PMID:1740468; http://dx.doi.org/10.1083/jcb.116.5.1095
- Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 1998; 140:1285-95; PMID:9508763; http://dx.doi.org/10.1083/jcb.140.6.1285
- Maya-Mendoza A, Olivares-Chauvet P, Shaw A, Jackson DA. S phase progression in human cells is dictated by the genetic continuity of DNA foci. PLoS Genet 2010; 6:e1000900; PMID:20386742; http://dx.doi.org/10.1371/journal.pgen.1000900
- Ma H, Samarabandu J, Devdhar RS, Acharya R, Cheng PC, Meng C, Berezney R. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J Cell Biol 1998; 143:1415-25; PMID:9852140; http://dx.doi.org/10.1083/jcb.143.6.1415
- Sparvoli E, Levi M, Rossi E. Replicon clusters may form structurally stable complexes of chromatin and chromosomes. J Cell Sci 1994; 107(Pt 1):3097-103; PMID:7699008
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 2010; 38:603-13; PMID:20513434; http://dx.doi.org/10.1016/j.molcel.2010.03.016
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012; 485:376-80; PMID:22495300; http://dx.doi.org/10.1038/nature11082
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014; 515:402-5; PMID:25409831; http://dx.doi.org/10.1038/nature13986
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang C-W, Lyou Y, Townes TM, Schübeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 2008; 6:e245; PMID:18842067; http://dx.doi.org/10.1371/journal.pbio.0060245
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. Chromatin architecture reorganization during stem cell differentiation. Nature 2015; 518:331-6; PMID:25693564; http://dx.doi.org/10.1038/nature14222
- Dileep V, Ay F, Sima J, Vera DL, Noble WS, Gilbert DM. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res 2015; 25:1104-13; PMID:25995270; http://dx.doi.org/10.1101/gr.183699.114
- Lange C, Calegari F. Cdks and cyclins link G1 length and differentiation of embryonic, neural and hematopoietic stem cells. Cell cycle Georg Tex 2010; 9:1893-900; PMID:20436288; http://dx.doi.org/10.4161/cc.9.10.11598
- Calder A, Roth-Albin I, Bhatia S, Pilquil C, Lee JH, Bhatia M, Levadoux-Martin M, McNicol J, Russell J, Collins T, et al. Lengthened G1 phase indicates differentiation status in human embryonic stem cells. Stem Cells Dev 2013; 22:279-95; PMID:22827698; http://dx.doi.org/10.1089/scd.2012.0168
- Becker KA, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Human embryonic stem cells are pre-mitotically committed to self-renewal and acquire a lengthened G1 phase upon lineage programming. J Cell Physiol 2010; 222:103-10; PMID:19774559; http://dx.doi.org/10.1002/jcp.21925
- White J, Dalton S. Cell cycle control of embryonic stem cells. Stem Cell Rev 2005; 1:131-8; PMID:17142847; http://dx.doi.org/10.1385/SCR:1:2:131
- Gilbert DM. Cell fate transitions and the replication timing decision point. J Cell Biol 2010; 191:899-903; PMID:21115801; http://dx.doi.org/10.1083/jcb.201007125
- Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol 2006; 7:540-6; PMID:16723974; http://dx.doi.org/10.1038/nrm1938
- Panning MM, Gilbert DM. Spatio-temporal organization of DNA replication in murine embryonic stem, primary, and immortalized cells. J Cell Biochem 2005; 95:74-82; PMID:15723284; http://dx.doi.org/10.1002/jcb.20395
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008; 132:487-98; PMID:18267078; http://dx.doi.org/10.1016/j.cell.2007.12.033
- Singh AM, Chappell J, Trost R, Lin L, Wang T, Tang J, Matlock BK, Weller KP, Wu H, Zhao S, et al. Cell-Cycle Control of Developmentally Regulated Transcription Factors Accounts for Heterogeneity in Human Pluripotent Cells. Stem Cell Reports 2014; 2:398; PMID:24371808; http://dx.doi.org/10.1016/j.stemcr.2014.02.009
- Kuipers MA, Stasevich TJ, Sasaki T, Wilson KA, Hazelwood KL, McNally JG, Davidson MW, Gilbert DM. Highly stable loading of Mcm proteins onto chromatin in living cells requires replication to unload. J Cell Biol 2011; 192:29-41; PMID:21220507; http://dx.doi.org/10.1083/jcb.201007111
- Sasaki T, Li A, Gillespie PJ, Blow JJ, Gilbert DM. Evidence for a mammalian late-G1 phase inhibitor of replication licensing distinct from geminin or Cdk activity. Nucleus 2:455-64; PMID:21983086; http://dx.doi.org/10.4161/nucl.2.5.17859
- Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, Cardoso MC. Dynamics of DNA replication factories in living cells. J Cell Biol 2000; 149:271-80; PMID:10769021; http://dx.doi.org/10.1083/jcb.149.2.271
- Becker KA, Ghule PN, Therrien JA, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J Cell Physiol 2006; 209:883-93; PMID:16972248; http://dx.doi.org/10.1002/jcp.20776
- Schulz TC, Young HY, Agulnick AD, Babin MJ, Baetge EE, Bang AG, Bhoumik A, Cepa I, Cesario RM, Haakmeester C, et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS One 2012; 7:e37004; PMID:22623968; http://dx.doi.org/10.1371/journal.pone.0037004
- Loh KM, Ang LT, Zhang J, Kumar V, Ang J, Auyeong JQ, Lee KL, Choo SH, Lim CYY, Nichane M, et al. Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 2014; 14:237-52; PMID:24412311; http://dx.doi.org/10.1016/j.stem.2013.12.007
- Qu X-B, Pan J, Zhang C, Huang S-Y. Sox17 facilitates the differentiation of mouse embryonic stem cells into primitive and definitive endoderm in vitro. Dev Growth Differ 2008; 50:585-93; PMID:19238729; http://dx.doi.org/10.1111/j.1440-169X.2008.01056.x
- Wang P, Rodriguez RT, Wang J, Ghodasara A, Kim SK. Targeting SOX17 in human embryonic stem cells creates unique strategies for isolating and analyzing developing endoderm. Cell Stem Cell 2011; 8:335-46; PMID:21362573; http://dx.doi.org/10.1016/j.stem.2011.01.017
- Kanai-Azuma M, Kanai Y, Gad JM, Tajima Y, Taya C, Kurohmaru M, Sanai Y, Yonekawa H, Yazaki K, Tam PPL, et al. Depletion of definitive gut endoderm in Sox17-null mutant mice. Development 2002; 129:2367-79; PMID:11973269
- Borghese L, Dolezalova D, Opitz T, Haupt S, Leinhaas A, Steinfarz B, Koch P, Edenhofer F, Hampl A, Brüstle O. Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 2010; 28:955-64; PMID:20235098; http://dx.doi.org/10.1002/stem.408
- Manders EMM, Kimura H, Cook PR. Direct Imaging of DNA in Living Cells Reveals the Dynamics of Chromosome Formation. J Cell Biol 1999; 144:813-22; PMID:10085283; http://dx.doi.org/10.1083/jcb.144.5.813
- Sela Y, Molotski N, Golan S, Itskovitz‐Eldor J, Soen Y. Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of pRB. Stem cells Dayt Ohio 2012; 30:1097-108; http://dx.doi.org/10.1002/stem.1078
- Pauklin S, Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell 2013; 155:135-47; PMID:24074866; http://dx.doi.org/10.1016/j.cell.2013.08.031
- Pauklin S, Madrigal P, Bertero A, Vallier L. Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes Dev 2016; 30:421-33; PMID:26883361; http://dx.doi.org/10.1101/gad.271452.115
- Singh AM, Chappell J, Trost R, Lin L, Wang T, Tang J, Wu H, Zhao S, Jin P, Dalton S. Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells. Stem Cell Rep 2013; 1:532-44; PMID:24371808; http://dx.doi.org/10.1016/j.stemcr.2013.10.009
- Edenberg HJ, Huberman JA. Eukaryotic chromosome replication. Annu Rev Genet 1975; 9:245-84; PMID:55095; http://dx.doi.org/10.1146/annurev.ge.09.120175.001333
- Hand R. Eucaryotic DNA: organization of the genome for replication. Cell 1978; 15:317-25; PMID:719745; http://dx.doi.org/10.1016/0092-8674(78)90001-6
- Ma H, Siegel AJ, Berezney R. Association of chromosome territories with the nuclear matrix. J Cell Biol 1999; 146:531-42; PMID:10444063; http://dx.doi.org/10.1083/jcb.146.3.531
- Berezney R, Dubey DD, Huberman JA. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000; 108:471-84; PMID:10794569; http://dx.doi.org/10.1007/s004120050399
- Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC. DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol Cell 2002; 10:1355-65; PMID:12504011; http://dx.doi.org/10.1016/S1097-2765(02)00729-3
- Dileep V, Rivera-Mulia JC, Sima J, Gilbert DM. Large-scale chromatin structure-function relationships during the cell cycle and development: insights from replication timing. Cold Spring Harb Symp Quant Biol 2015; PMID:26590169
- Tsubouchi T, Soza-Ried J, Brown K, Piccolo FM, Cantone I, Landeira D, Bagci H, Hochegger H, Merkenschlager M, Fisher AG. DNA synthesis is required for reprogramming mediated by stem cell fusion. Cell 2013; 152:873-83; PMID:23415233; http://dx.doi.org/10.1016/j.cell.2013.01.012
- Shachar S, Voss TC, Pegoraro G, Sciascia N, Misteli T. Identification of gene positioning factors using high-throughput imaging mapping. Cell 2015; 162:911-23; PMID:26276637; http://dx.doi.org/10.1016/j.cell.2015.07.035
- Pop R, Shearstone JR, Shen Q, Liu Y, Hallstrom K, Koulnis M, Gribnau J, Socolovsky M. A key commitment step in erythropoiesis is synchronized with the cell cycle clock through mutual inhibition between PU.1 and S-phase progression. PLoS Biol 2010; 8:e1000484; PMID:20877475; http://dx.doi.org/10.1371/journal.pbio.1000484
- Zhang Z, Shibahara K, Stillman B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 2000; 408:221-5; PMID:11089978; http://dx.doi.org/10.1038/35048530
- Shibahara K, Stillman B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999; 96:575-85; PMID:10052459; http://dx.doi.org/10.1016/S0092-8674(00)80661-3