
ABSTRACT
PTEN functions as a guardian of the genome through multiple mechanisms. We have previously established that PTEN maintains the structural integrity of chromosomes. In this report, we demonstrate a fundamental role of PTEN in controlling chromosome inheritance to prevent gross genomic alterations. Disruption of PTEN or depletion of PTEN protein phosphatase activity causes abnormal chromosome content, manifested by enlarged or polyploid nuclei. We further identify polo-like kinase 1 (PLK1) as a substrate of PTEN phosphatase. PTEN can physically associate with PLK1 and reduce PLK1 phosphorylation in a phosphatase-dependent manner. We show that PTEN deficiency leads to PLK1 phosphorylation and that a phospho-mimicking PLK1 mutant causes polyploidy, imitating functional deficiency of PTEN phosphatase. Inhibition of PLK1 activity or overexpression of a non-phosphorylatable PLK1 mutant reduces the polyploid cell population. These data reveal a new mechanism by which PTEN controls genomic stability during cell division.
Introduction
PTEN is one of the most frequently mutated genes in human cancer, and chromosome instability (CIN) is often found in cells and tissues lacking PTEN function.Citation1-4 PTEN targets phosphatidylinositol-3,4,5-triphosphate (PIP3) and antagonizes the major survival pathway regulated by phosphatidylinositol 3-kinase (PI3K)/Akt.Citation5-7 Increasing evidence indicates that nuclear PTEN has fundamental functions in maintenance of chromosomal stability,Citation2,8-14 suggesting there are as yet unidentified mechanisms in the process of chromosome inheritance for which the function of PTEN is indispensable. It has been shown that PTEN plays an essential role in maintaining the structural integrity of individual chromosomes,Citation2 which supports recognition of PTEN as a guardian of the genome.Citation15,16 Although PTEN mutations and aneuploidy are concurrent events in tumorigenesis,Citation1,2,17,18 it remains elusive how PTEN controls numerical stability of chromosome transmission.
Aneuploidy is a common feature of cancer cells that is often preceded by an intermediate state of tetraploidy or polyploidy.Citation19-21 To explore the molecular mechanisms that bring about polyploidization, a genetic screen in yeast has identified 39 genes essential for the viability of polyploid yeast. Almost all of these genes are involved in mitosis, encoding components in mitotic spindles, sister chromatid cohesion or homologous recombination.Citation22 Polo-like kinase 1 (PLK1) is the vertebrate homolog of Polo in Drosophila,Citation23 of Plo1 in fission yeastCitation24 and of Cdc5 in budding yeast.Citation25 PLK1 is an essential mitotic kinase that controls spindle bipolarity,Citation23 mitotic entryCitation26 and mitotic exit,Citation27 as well as cytokinesis.Citation28 Recent studies have demonstrated PLK1 functions in late mitosis in controlling cleavage furrow formation in anaphase cells.Citation29-31 Deregulation of PLK1 causes various forms of mitotic defects and leads to karyotypic instability, which may contribute to oncogenic transformation.Citation32 For example, constitutively active Xenopus Plx1 induces formation of multiple nucleiCitation33; aberrant Cdc5 leads to premature separation of sister chromatidsCitation34; and Drosophila polo mutants exhibit multinucleation due to failure of cytokinesis.Citation35 Moreover, there is significant correlation of PLK1 overexpression with tumor metastatic potentialCitation36 and with prognosis in cancer patients.Citation37-40 PLK1 can be phosphorylated by protein kinase A, Cdc2,Citation41,42 and aurora A,Citation43,44 and phosphorylation of PLK1 at Thr210 has been identified as a major event in the activation of PLK1.Citation41,45
Here we report that PTEN protects the genome by preventing whole chromosome instability, which represents a novel function of PTEN in tumor suppression. In addition, the prototypical vertebrate polo kinase PLK1 emerges in this study as a protein substrate of PTEN phosphatase, and we show that PTEN is capable of physically interacting with PLK1 and dephosphorylating this critical mitotic kinase. On the basis of these findings PTEN may be characterized as a mitotic phosphatase that regulates PLK1 and prevents polyploidy. Our data emphasize the importance of the balance between mitotic kinases and phosphatases for the fidelity of chromosome inheritance during cell division.
Results
Loss of PTEN leads to spontaneous polyploidy and resistance to spindle disruption
Signature genetic alterations associated with Pten disruption include segmental chromosome aberrations such as translocations and chromosomal breakage.Citation2,46-48 PTEN-deficient cells also spontaneously exhibit aneuploidy,Citation1,2 reflecting whole chromosome instability. In order to determine whether Pten deletion can induce gross alteration of chromosome content, we compared cell cycle profiles and DNA ploidy status in Pten+/+ and Pten−/− mouse embryonic fibroblasts (MEFs). As shown in , Pten−/− MEFs accumulate either as a hypodiploid (<2N) or polyploid population (>4N), in association with a dramatic reduction in the cycling cell population (2N-4N) as compared with wild-type cells (62.2 ± 5.1% vs. 83.5 ± 1.1%, ). These data indicate that Pten depletion induces spontaneous formation of polyploid cells. Faulty spindle production from exogenous stimuli may exacerbate these spontaneous aberrations. To test this idea, we challenged Pten proficient and deficient cells with nocodazole, a microtubule-depolymerizing reagent, prior to evaluation of polyploidy status. The polyploid population in Pten null cells is further increased (51.9 ± 4.7% vs. 28.5 ± 2.2%, ), whereas the ploidy status remains much more stable in Pten+/+ MEFs. The challenge of spindle disruption clearly exacerbates the already unstable karyotype in Pten null cells by further reducing the cycling population associated with aggravation of polyploidization. To determine how these polyploid cells respond to spindle perturbation in terms of ultimate cell fate, we monitored cell survival of Pten+/+ and Pten−/− MEFs in the presence of nocodazole. Nocodazole diminishes survival of Pten+/+ cells to a significantly greater extent than that of Pten−/− cells and Pten null cells appear to be more resistant to this agent (). These results suggest that loss of Pten confers cellular resistance to spindle-disrupting drugs.
Figure 1. Pten disruption results in polyploidy and nocodazole resistance. (A and B) Flow cytometry analysis of ploidy distribution and DNA content in Pten+/+ and Pten−/− MEFs. Data are presented as mean ± SEM of 3 independent experiments (*, p < 0.05; as compared with Pten+/+ cells with the same ploidy population). (C) Karyotypic profiles of Pten+/+ and Pten−/− MEFs with or without spindle disruption by nocodazole (1.5 μM). Data are summarized from 3 independent experiments and analyzed with the paired t-test. *, p < 0.05; **, p < 0.01; as compared with corresponding untreated or nocodazole treated Pten+/+ cells. (D) Growth curves for Pten+/+ and Pten−/− MEFs in response to nocodazole (1.5 μM). (E) HeLa cells were transfected with pSuper/sh PTEN and PTEN expression was evaluated by Western blotting and compared with cells containing a control pSuper/sh control vector. (F) Immunofluorescent staining of microtubules using an anti-α-tubulin antibody and visualized with an FITC-conjugated species-matched secondary antibody. White arrowheads and red arrows indicate giant cells containing a single enlarged nucleus and multiple nuclei respectively. (G) A representative comparison of cell cycle and ploidy profiles in thymidine synchronized HeLa cells containing PTEN shRNA or scrambled shRNA, at a time point 12 hours after release from double-thymidine block (DTB). (H) Survival curves for PTEN-knockdown and control HeLa cells treated with 1.5 μM nocodazole for 24 h and 48 h.
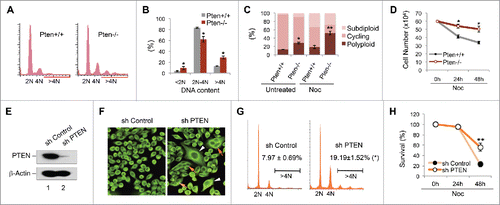
To consolidate the data obtained from this mouse cell system, we knocked down PTEN in HeLa cells by PTEN shRNA (). As compared with cells containing scrambled control shRNA, the effect of PTEN silencing is dramatic with generation of numbers of giant cells (), including multinucleated cells and cells with a single enlarged nucleus. We used a double-thymidine block (DTB) synchronization approach to examine the cell cycle profile following PTEN knockdown. Flow cytometric analysis revealed a consistent increase of the polyploid population in cells containing PTEN shRNA at each time point during the 0∼16-hour monitoring process following DTB release. The most marked difference of polyploidy frequency between PTEN knockdown cells and control cells occurs following mitosis (12∼16 hours after DTB release). At the 12-hour time point, there is a 2.4-fold increase in polyploid DNA content in PTEN knockdown cells as compared with control cells (). These results suggest that the aberrant chromosome complement associated with PTEN knockdown may result from chromosomal alterations generated during cell division. In evaluation of cell sensitivity to spindle pertubation, we found that PTEN knockdown cells are more resistant to nocodazole treatment (). These data collectively demonstrate that PTEN deficiency results in cellular resistance to spindle damage, which may result from defective mitosis and polyploidization.
In order to determine whether PTEN deficiency causes mitotic defects, we employed time-lapse microscopy to monitor the dynamics of chromosome segregation and compare live cell progression through mitosis using cells expressing a GFP-labeled chromatin marker, histone H2B-GFP. Failure of chromosome segregation and/or cytokinesis is a prominent phenomenon in cells lacking PTEN. For example, cells containing PTEN shRNA may arrest in metaphase for hours without initiating anaphase, and chromosomes align normally on the metaphase plate but fail to segregate (Fig. S1, middle panel). PTEN knockdown cells often exhibit chromosome non-disjunction or cleavage furrow regression, leading to failure of cytokinesis (Fig. S1, lower panel). Therefore, deficiency of PTEN causes erroneous chromosome segregation or incomplete cytokinesis, leading to polyploidization.
Polo-like kinase 1 is a PTEN-associated protein during mitosis
The mitotic and karyotypic abnormalities following PTEN depletion suggest that PTEN may be physically involved in mitotic control. In order to identify potential cellular factors in the PTEN mitotic pathway, we used a biochemical purification approach to isolate proteins that specifically interact with PTEN. A stable HeLa cell line expressing FLAG-HA double-tagged PTEN () was employed for tandem affinity purification of potential PTEN-associated proteins from mitosis-enriched cells. Mass spectrometric analysis of a ∼70 kD band identified polo-like kinase 1 (PLK1) as a potential PTEN-interacting protein ( and S2).
Figure 2. PLK1 is a PTEN-associated mitotic protein. (A) Expression of FH-PTEN and endogenous PTEN in FH-PTEN-expressing HeLa cells. Western blot analysis of HeLa cells containing FH-PTEN or a control vector with an anti-PTEN (upper panel) or anti-FLAG (lower panel) antibody. (B) Identification of Plk1 as a component of PTEN-associated protein complexes by pull-down assay. PTEN-complexes from HeLa/FLAG-HA-PTEN cells and a control elution from HeLa cells containing an empty vector were separated on SDS-PAGE following tandem affinity purification. A ∼70-kDa band was analyzed by mass-spectrometry and revealed PLK1 as a potential PTEN-associated protein. (C) Direct interaction of PTEN with PLK1 in vitro. Sf9 cells were infected with indicated recombinant baculovirus for 72 hours and subsequently harvested for protein purification using either an Ni-NTA agarose column or an anti-FLAG M2 affinity gel. Purified His-tagged proteins were incubated with purified FLAG-PLK1 protein followed by precipitation with Ni-NTA beads. Samples were then subjected to analysis of FLAG-PLK1 expression (upper panel). His-tagged PTEN and control protein in these reactions were shown in the lower panel. (D) Physical association of endogenous PTEN and PLK1. HeLa cells as well as MEFs with or without Pten deletion were subjected to nocodazole treatment (50 nM, 8 h) followed by immunoprecipitation with a rabbit anti-PTEN antibody and subsequent evaluation for PLK1 and PTEN by Western blotting. An isotype-matched mouse IgG was used as a control for immunoprecipitation and ExactaCruz reagents were used to reduce the interference of IgG bands. (E) Cell cycle-dependent PTEN-PLK1 association during cell division. HeLa cells were synchronized by DTB and released after different times as indicated. The interaction between PTEN and PLK1 was examined by PTEN immunoprecipitation for detection of PTEN-associated PLK1 by Western analysis. Cyclin B expression in synchronized pre-IP samples is shown to indicate mitotic entry and exit.
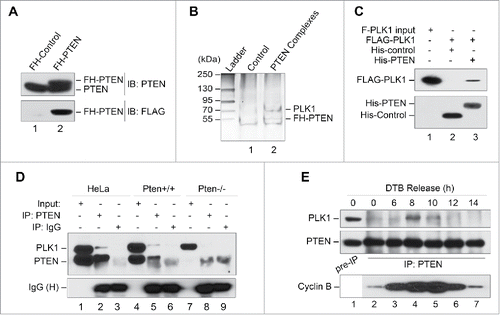
In order to validate the pull-down results, we performed an in vitro binding assay using purified recombinant FLAG-tagged PLK1 (FLAG-PLK1) and His-tagged PTEN (His-PTEN). As expected, FLAG-PLK1 associates with His-PTEN but not with an irrelevant protein His-control (), indicating that PTEN can directly interact with PLK1. We next examined the in vivo interaction between PTEN and PLK1. As shown in , endogenous PLK1 in HeLa cells is detectable in PTEN immunoprecipitates, but not in immunoprecipitates of a species-matched non-specific IgG (lane 2 vs. lane 3). Similarly, physical association of Pten with Plk1 can be found in Pten+/+, but not in Pten−/− MEFs (). These data suggest that PTEN is physically associated with PLK1.
To further delineate the relationship between PTEN and PLK1, we examined their interaction in the cell cycle particularly during mitosis. DTB synchronization and co-immunoprecipitation experiments reveal that PTEN primarily interacts with PLK1 during mitosis (). This interaction peaks when cells enter mitosis (8 h following DTB) and subsequently decreases as cells exit mitosis. These results depict a dynamic interaction between PTEN and PLK1, which implies they have a functional interplay in mitosis. Therefore, it was next imperative to determine the mechanism by which PTEN interacts with PLK1 in the control of chromosome inheritance during cell division.
PTEN deficiency results in PLK1 hyperphosphorylation
PLK1 is activated by phosphorylation on residue Thr210 within its activation loop.Citation49 In order to delineate a signaling link between PTEN and PLK1, we first examined the phosphorylation status of Plk1 in Pten-deficient cells, and found that the level of Plk1 phosphorylation at Thr210 is markedly increased in Pten−/− MEFs as compared to Pten+/+ MEFs in both the presence and absence of nocodazole treatment (). We also observed an elevation of the total level of Plk1 expression in Pten null cells, which may result from impairment of APC-Cdh1-mediated Plk1 degradation.Citation8 Nevertheless, the expression change is less significant as compared with that of phosphorylation levels, suggesting that Pten may directly target PLK1 to regulate its phosphorylation.
Figure 3. PLK1 is a protein substrate of PTEN phosphatase. (A and B) Pten depletion induces Plk1 phosphorylation. Pten+/+ and Pten−/− MEFs cultured with or without nocodazole (50 nM, 8 h, A), as well as various tissues from mice with wild-type Pten or heterozygous Pten deletion (B), were analyzed for Plk1 phosphorylation and abundance by immunoblotting with site-specific phospho-Plk1 (Thr210) and total Plk1 antibodies. β-Actin was used as a loading control. (C) PTEN directly and dose-dependently dephosphorylates PLK1. Sf9-expressed His-PTEN protein was purified using a Ni-NTA agarose column. FLAG-PLK1 expressed in Sf9 cells was immunoprecipitated with anti-FLAG M2 beads and incubated with increasing amounts of His-PTEN proteins, followed by Western blot analysis of PLK1 phosphorylation. The same blot was probed with PLK1 antibody to show PLK1 levels loaded in each lane. (D) Ectopic PTEN suppresses PLK1 kinase activity. Phosphorylation of NudC, a substrate of PLK1 kinase, was analyzed by immunoblotting in Pten−/− MEFs containing ectopic PTEN in the presence and absence of PLK1T210D, an enzymatically active form of PLK1.
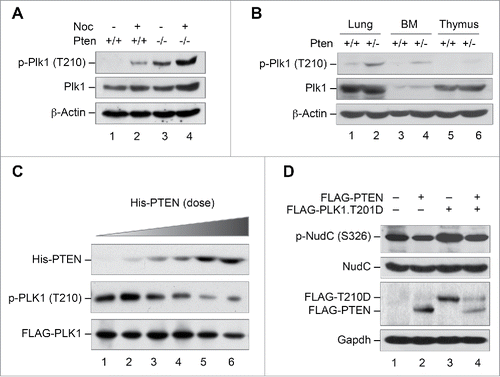
In mice, decreasing Pten dosage correlates with increasing tumor susceptibility,Citation50,51 suggesting sufficient levels of Pten are required for suppressing tumorigenesis. In order to determine whether Pten haploinsufficiency also affects PLK1 phosphorylation, we examined the status of PLK1 phosphorylation in various tissues from mice with heterozygous Pten deletion. As expected, PLK1 is phosphorylated in various Pten+/− tissues including lung, bone marrow and thymus, as compared with wild-type tissues from littermate control mice (). These results suggested that disruption of Pten is associated with spontaneous hyperphosphorylation of PLK1.
PTEN directly dephosphorylates PLK1 and reduces its kinase activity
Because PTEN regulates cellular processes through its dual phosphatase activity,Citation52,53 PTEN may employ its protein phosphatase function to dephosphorylate and inactivate PLK1. To determine whether PTEN can directly dephosphorylate PLK1 in vitro, we employed a phosphatase assay to evaluate PLK1 phosphorylation following incubating purified PLK1 protein with increasing amounts of His-tagged PTEN. As shown in , PLK1 phosphorylation is reduced by PTEN in a dose-dependent manner. In contrast, the total level of PLK1 remains relatively stable although a slight reduction is noticeable in the presence of very high concentrations of PTEN. These data suggest that PLK1 can serve as a direct substrate of PTEN phosphatase and that exhaustive dephosphorylation of PLK1 by PTEN may result in a reduction of the PLK1 total expression level.
Since PLK1 phosphorylation at the T210 residue is required for its kinase activity, one would expect that PTEN regulation of PLK1 phosphorylation might affect its downstream targets. Nuclear distribution gene C (NudC) is a substrate of PLK1 kinase that mediates PLK1 function in cytokinesis,Citation54 which may also serve as a target of the PTEN-PLK1 signaling axis in preventing cytokinesis failure and polyploidy. We therefore chose NudC to assess how NudC phosphorylation is influenced by the interaction between PTEN and PLK1. To this end, we constructed a phospho-mimetic PLK1 plasmid, PLK1T210D, to imitate enzymatically active PLK1.Citation49 NudC phosphorylation was then examined in Pten null MEFs transfected with PTEN and PLK1T210D individually and in combination. Pten null cells express a high basal level of NudC phosphorylation, likely due to PLK1 activation. Additional PLK1 activation by ectopic PLK1T210D further elevates NudC phosphoryolation only slightly. Nevertheless, NudC phosphorylation is reduced by ectopic PTEN, both alone and in combination with PLK1T210D (). These results suggest that PTEN can antagonize the kinase activity of both endogenous and enzymatically active PLK1, likely through a dephosphorylation mechanism.
PTEN inhibits PLK1 in a phosphatase-dependent manner
To further validate the role of PTEN phosphatase activity in suppression of PLK1 phosphorylation and in protection against polyploidization, we examined the level of Plk1 phosphorylation in Pten null cells with ectopic expression of wild-type PTEN or 2 phosphatase-deficient PTEN mutants. PTEN is well known to antagonize the PI3K-mediated lipid pathway.Citation5 In addition, PTEN can also act as a protein phosphatase to dephosphorylate protein substrates.Citation55,56 To distinguish its activities in suppressing the lipid or the protein pathways, 2 PTEN mutants have been commonly used. PTENC124S lacks both lipid and protein phosphatase activity,Citation57 whereas PTENG129E is lipid phosphatase deficient but retains protein phosphatase activity.Citation58 Plk1 phosphorylation is markedly elevated in Pten null cells, as compared with Pten+/+ MEFs (). Introduction of wild-type PTEN significantly reduces Plk1 phosphorylation, as compared with Pten−/− cells transfected with a control vector. Reduction of Plk1 phosphorylation mirrors the change in Akt phosphorylation (), suggesting that Plk1 serves as a target for Pten in the nucleus in a manner similar to that in which Akt acts as a signaling target of Pten in the cytoplasm. However, phosphorylation of Plk1 at Thr210 remains at a high level in cells transfected with the PTEN mutant PTENC124S, which is deficient in both protein and lipid phosphatase activity (). In contrast, the lipid phosphatase-deficient PTEN mutant PTENG129E still suppresses Plk1 phosphorylation to an extent comparable to wild-type PTEN (). It is of particular interest to compare the phosphorylation changes in Plk1 () with those of Akt () in response to ectopic expression of different mutant forms of PTEN. Although both PTENC124S and PTENG129E lack ability to inhibit Akt phosphorylation (), the lipid phosphatase-deficient form of PTEN, PTENG129E, retains its ability to suppress Plk1 phosphorylation (). These data indicate that suppression of Plk1 phosphorylation by PTEN requires its protein phosphatase activity.
Figure 4. PTEN reduces PLK1 phosphorylation, PLK1 stability and polyploidy in a phosphatase-dependent manner. (A) Pten−/− MEFs were transfected with vectors encoding FLAG-tagged wild-type PTEN or the phosphatase-deficient PTEN mutants PTENC124S and PTENG129E, and subjected to immunoprecipitation with anti-Plk1 antibody followed by evaluation for phospho-PLK1 (Thr210). The expression of PLK1 and β-actin was then evaluated by re-blotting with corresponding antibodies. (B) Cell lysates identical to those in (A) prior to immunoprecipitation were processed for immunoblot analysis of phospho-Akt (Ser473). Endogenous and ectopic FLAG-PTEN expression was evaluated using anti-PTEN and anti-FLAG monoclonal antibodies respectively. Equal protein loading was demonstrated by re-probing the same blot with anti-β-actin antibody. (C) Pten null MEFs were co-transfected with FLAG-tagged PLK1 and different forms of PTEN (wild-type or phosphatase-deficient mutants) prior to analysis of FLAG-PLK1 expression. (D) Pten+/+ MEFs as well as Pten−/− MEFs containing wild-type PTEN, or one of the 2 phosphatase-deficient mutants (PTENC124S and PTENG129E) were cultured for 48 h followed by flow cytometric analyis of ploidy status and analyzed with the paired t-test for statistical significance. *, p < 0.05; **, p < 0.01. (E) Pten−/− MEFs containing wild-type PTEN, PTENC124S or PTENG129E were treated with 1.5 μM nocodazole for 24 h and subjected to survival analysis by trypan blue exclusion. *, p < 0.05.
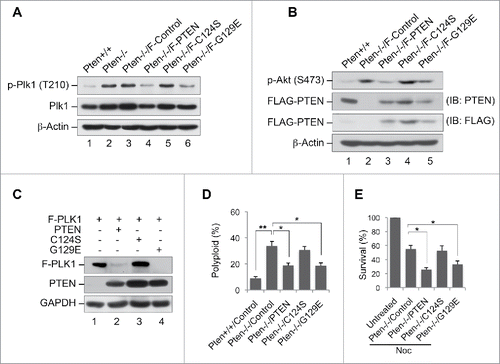
Phosphorylation is associated with protein stability during cell division.Citation59 In agreement with this notion, we often observed a concomitant change of PLK1 expression in Pten-deficient cells that corresponds to its phosphorylation state (see ). We therefore hypothesized that dephosphorylation of PLK1 by PTEN may render a reduction of PLK1 stability. To test this hypothesis, we co-transfected Pten null cells with FLAG-tagged PLK1 and different forms of PTEN with or without phosphatase activity prior to analysis of FLAG-PLK1 expression. As shown in , the expression of FLAG-PLK1 is significantly diminished by wild-type PTEN but not by PTENC124S, the mutant lacking both protein and lipid phosphatase activity. The PTENG129E mutant lacking lipid phosphatase activity, however, retains the capability of reducing FLAG-PLK1 expression (). These results support that PTEN reduces the expression level of ectopic FLAG-PLK1 in a manner similar to PTEN suppression of PLK1 phosphorylation (), suggesting that PLK1 phosphorylation plays an important role in maintaining its protein stability. These data highlight a phosphatase-dependent regulation of PLK1 by PTEN leading to a successive inhibition of PLK1 phosphorylation and stability.
Phosphatase function of PTEN is essential for prevention of polyploidization and restoration of sensitivity to spindle drugs
In order to determine whether differences in PTEN phosphatase activity are reflected in phenotypic changes, we investigated ploidy status in Pten−/− cells ectopically expressing PTEN with differing phosphatase activity (wild-type PTEN, PTENC124S or PTENG129E). As shown in , ectopic expression of wild-type PTEN as well as the lipid-phosphatase-deficient PTEN mutant (PTENG129E) results in a dramatic reduction of the polyploid population. However, the PTENC124S mutant which lacks both lipid and protein phosphatase activity fails to cause such a reduction.
PTEN deficiency not only induces ploidy aberration but also conveys resistance to spindle disruption (). If polyploidy were functionally linked to cellular resistance to spindle disruption, one would expect that amelioration of karyotypic instability would restore cellular sensitivity to spindle poison. As expected, a significantly greater cell death response to nocodazole is induced by ectopic expression of wild-type PTEN as well as by PTENG129E, but not by PTENC124S (). Therefore, PTEN's function in sensitizing cells to spindle disruption is dependent upon its protein phosphatase activity, in a manner parallel to PTEN's effect on ploidy status. These results indicate that reconstitution of PTEN expression in Pten null cells can restore the ability of PTEN phosphatase to suppress Plk1 phosphorylation and in consequence, reduce polyploidy and restore cellular sensitivity to spindle disruption. The phosphatase activity of PTEN is therefore necessary not only for counterbalancing PLK1 phosphorylation and stability but also for maintaining normal chromosomal content and cellular responsiveness to spindle damage.
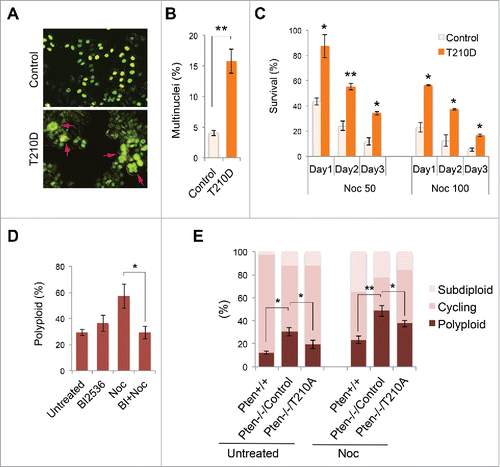
Constitutive activation of PLK1 mimics PTEN deficiency to induce polyploidy whereas inhibition of Plk1 ameliorates karyotypic instability
In order to investigate the functional interplay between PTEN and PLK1, we employed 2 strategies to determine how PLK1 phosphorylation contributes to an aberrant chromosome complement. First, we sought to determine whether abnormal PLK1 activation mimics PTEN deficiency and how the phospho-mimicking mutant PLK1T210D affects ploidy status and cellular sensitivity to spindle poison. Ectopic expression of constitutively activated PLK1 results in significantly increased numbers of multinucleated cells (), indicating that PLK1T210D induces polyploidization. Moreover, the phospho-mimicking PLK1T210D endows cells with resistance to spindle disruption by nocodazole (). All of these phenotypic features induced by PLK1T210D mirror our observations in PTEN-deficient cells (), suggesting that PLK1 hyperphosphorylation may mediate generation of polyploidy following PTEN depletion. To consolidate these observations, we next investigated whether chemical inhibition of PLK1 activity reduces polyploidy in Pten-deficient cells. We first utilized BI 2536, a specific Plk1 inhibitor Citation60,61, to block Plk1 activity in Pten null cells. As shown in , the polyploid population in Pten null cells is increased by nocodazole, and this effect can be blocked by pretreatment with BI 2536. Moreover, overexpression of a non-phosphorylatable PLK1T210A mutant in Pten null cells can ameliorate the abnormal karyotype, as demonstrated by an increase in the cycling diploid cell population and reduction in numbers of polyploid cells in both the presence and absence of nocodazole treatment (). These data indicate that inhibition of PLK1 can correct polyploidy in Pten null cells and restore normal karyotypic characteristics. Our data thus suggest that deregulation of PLK1 activity is responsible for polyploidization in Pten-deficient cells. Therefore, the functional interplay between a potent tumor suppressor phosphatase (PTEN) and a key mitotic kinase (PLK1) may serve to prevent polyploidization and maintain chromosomal stability.
Figure 5. Constitutively active PLK1 induces multinucleation whereas inhibition of PLK1 mitigates polyploidy. (A) HeLa cells were co-transfected with GFP-labeled histone H2B as well as PLK1T210D or a control expression vector, followed by immunofluorescent microscopic analysis of cell morphology. Arrows indicate multinucleated cells. (B) HeLa cells with or without PLK1T210D (n ≥ 400) were analyzed in randomly selected regions and the percentage of multinucleated cells was plotted on the histogram. **, p < 0.01. (C) Cells with or without PLK1T210D were treated with different doses of nocodazole and cell survival was examined by trypan blue exclusion at various times during a 3-day course. *, p < 0.05; **, p < 0.01; as compared with control cells analyzed on the same day. (D) Pten−/− MEFs were pre-treated with BI 2536 (100 nM, 30 min) prior to nocodazole treatment (1.5 μM) for 48 h. Cells were evaluated for polyploidization by flow cytometry. *, p < 0.05. (E) Pten−/− MEFs transiently transfected with PLK1T210A or an empty plasmid were used as controls and were treated with nocodazole (1.5 μM) for 48 h followed by ploidy analysis. *, p < 0.05; **, p < 0.01.
Discussion
In this study, we demonstrate that PTEN protects the genome against polyploidization through regulation of the mitotic kinase PLK1. Our data highlight the phosphatase function of PTEN that is required for maintaining the optimal phosphorylation and stability of PLK1 to prevent its overactivation. The fact that constitutively active PLK1 induces polyploidy whereas inhibition of PLK1 phosphorylation reduces the frequency of polyploidization suggests that aberrant PLK1 phosphorylation mediates polyploidization as a result of PTEN deficiency. These results corroborate that PTEN is essential for the maintenance of whole chromosome stability by tuning the balance of mitotic phosphorylation. Our findings thus reveal a mechanism for cell division that is based on the interplay of PTEN phosphatase and PLK1 kinase, and this mechanism is essential for faithful genetic transmission.
PLK1 is regulated in a meticulous manner to ensure optimal levels of expression and activity over various stages of the cell cycle. Although PLK1 activation by upstream kinases has been well studied,Citation43,44 how PLK1 is dephosphorylated remains obscure. Our study demonstrates that PTEN phosphatase directly interacts with PLK1 and inhibits its activity by dephosphorylation. Identification of PLK1 as a molecular target of the phosphatase PTEN advances current knowledge of PTEN function beyond its role as an antagonist of the PI3K/Akt pathway. The functional interplay between PTEN and PLK1 thus forms a control mechanism for maintaining genomic stability during cell division.
PLK1 controls spindle bipolarity Citation23 and precise progression of mitosis from entry Citation26 to exit.Citation27 By regulating such a versatile mitotic kinase, PTEN may also play multiple roles in various aspects of mitotic control. In addition, PTEN may regulate PLK1 through different mechanisms. It has been reported that PTEN regulates the APC-CDH1 complex to reduce PLK1 protein stability.Citation8 Moreover, elevated expression of PLK1 in PTEN-depleted cancer cells can confer the tumorigenic competence.Citation62 Interestingly, PTEN function in APC-CDH1-mediated regulation of PLK1 protein levels appears to be phosphatase-independent and involved in cellular senescence.Citation8 Therefore, PTEN may utilize different mechanisms to regulate PLK1 in multiple cellular processes. Distinct mechanisms point to the same direction, e.g. PTEN suppresses PLK1 to ensure optimal expression levels and kinase activity of PLK1 during the cell cycle. PTEN phosphatase can directly inhibit PLK1 phosphorylation to reduce its stability whereas it can also influence PLK1 indirectly by targeting the APC-CDH1 complex. More interestingly, PTEN and PLK1 can mutually regulate each other as PLK1 can phosphorylate PTEN to regulate glucose metabolismCitation63 as well as mitotic timing.Citation64 The bidirectional interplay between PTEN and PLK1 may represent a signaling axis that plays a critical role in modulating cancer evolution. As the phospho-mimicking PLK1 mutant imitates PTEN deficiency and causes polyploidy, it is therefore intriguing to investigate whether PLK1 hyperphosphorylation may lead to tumorigenesis in future studies.
PTEN phosphatase may have different protein substrates necessary for its mitotic function. PLK1 may be only one among many nuclear PTEN targets that act to regulate the complicated process of cell division. For example, we have recently found that PTEN uses its protein phosphatase activity to regulate a critical mitotic motor protein (unpublished data). This in turn suggests PTEN acts as a calibrator of protein phosphorylation to maintain optimal activity of multiple mitotic proteins for normal well-orchestrated mitosis. It is of interest that the major cytoplasmic substrate of the PTEN lipid phosphatase PIP3 has recently been found in the midbody acting to control cytokinesis,Citation65 which argues PTEN is involved in the same process. As such, future efforts may uncover additional PTEN targets that together constitute a surveillance mechanism for ensuring mitotic fidelity.
Spindle perturbation is currently in use in clinical oncology as an anti-mitotic strategy which aims at activation of the spindle assembly checkpoint to cause mitotic arrest and subsequent cell death.Citation66 However, this strategy has limits as cancer cells may escape this type of cell death.Citation67 Our data similarly suggest that cells lacking PTEN may acquire a mechanism defective in the control of chromosome segregation resulting in polyploidy, which confers cellular resistance to anti-mitotic spindle drugs. Establishment of this novel link between functional deficiency of PTEN and resistance to spindle drugs provides a theoretical framework from which novel therapeutic approaches may be devised. For example, there is potential for restoration of cellular sensitivity to spindle drugs by inhibition of PLK1, and such a strategy could enhance chemotherapeutic efficiency by overcoming drug resistance caused by PTEN deficiency. Knowledge gained from this study will provide a new perspective for further mechanistic studies to identify additional intracellular targets for PTEN itself and PTEN-related drug development.
Materials and methods
Cells, tissues, antibodies, chemicals and plasmids
Pten+/+ and Pten−/− mouse embryo fibroblasts (MEFs) of less than 10 passages were used for phenotypic analyses. HeLa cells were from the American Type Culture Collection. Sf9 insect cells were from EMD Biosciences. Mammalian cells were cultured in MEM and Sf9 cells were cultured in Grace's insect medium, both supplemented with 10% fetal bovine serum (FBS). Monoclonal anti-PTEN antibodies were purchased from Santa Cruz biotechnology and Cascade Bioscience. A polyclonal PTEN antibody was generated by immunizing rabbits with a purified full-length PTEN protein. Anti-PLK1 and anti-phospho-PLK1 (Thr210) antibodies were from BioLegend. Nocodazole and BI 2536 were purchased from Sigma and AxonMedchem respectively.
shRNA and plasmids
The pSUPER RNAi system (Oligoengine Inc.) was used to construct PTEN-specific shRNA expression plasmids and scrambled control shRNA. The expression vectors containing FLAG or FLAG-HA tags were gifts from W. Gu.Citation68 The PLK1 expression plasmid was created by ligating the full-length coding region of human PLK1 into the expression vector with an N-terminal FLAG tag. Two PLK1 mutants, PLK1T210D and PLK1T210A, were generated by changing codon Thr210 from ACC to GAC or GCC respectively by using the QuikChange site-directed mutagenesis kit (Strategene) according to the manufacturer's protocol. Wild-type PTEN and phosphatase-deficient PTEN mutants, PTENC124S and PTENG129E, have been described previously.Citation2 A pFastBac1 expression vector (Invitrogen) was used to construct plasmids expressing PTEN and control proteins with His-tag.
Cell growth, cell cycle profile and ploidy analysis
Cells were plated so that 50% confluence was reached on the second day followed by continuing culture or nocodazole treatment with or without 30-min pretreatment with BI 2536 for an additional 48 h. Cells were then harvested as single cell suspension. Cell growth was analyzed with trypan blue exclusion. For cell cycle and ploidy profiles, cells were fixed with ice-cold 75% ethanol prior to staining with 4 μg/ml propidium iodide in PBS containing 0.1 mg/ml RNase A. DNA content of cells was analyzed using a Becton Dickinson FACScan flow cytometer, and the cell cycle distribution and ploidy status were determined using CellQuest Pro software. Three independent experiments were analyzed with the paired t-test and p < 0.05 was considered statistically significant.
Immunofluorescence, confocal microscopy and live cell imaging
Cells were fixed for immunofluorescence staining, confocal microscopy and live cell imaging. Exponentially growing cells were fixed with 4% formaldehyde in PHEM (60 mM Pipes, 25 mM Hepes, 10 mM EGTA, and 2 mM MgCl2, pH 6.9) and permeablized for immunostaining. Cell morphology was examined by staining microtubules and centrosomes with a monoclonal anti-α-tubulin (Sigma) antibody. Dynamics of cell cycle progression through mitosis were analyzed by time-lapse imaging using a Leica TCS SP5 confocal microscope. Image acquisition was performed with a 63× objective enclosed in a humidified incubation chamber with 5% CO2 at 37°C. Images were collected every 1.5–2.5 min for 5–20 hours. Images were analyzed by 3-dimensional maximum projection with LAS AF software.
FLAG-HA pull-down and mass spectrometry
To purify PTEN-associated mitotic proteins for mass spectrometric analysis, HeLa cells stably expressing FLAG-HA-tagged PTEN and control cells containing an empty FLAG-HA vector were treated with nocodazole (100 nM) for 8 hours. Nuclear fractions of cell extracts were subjected to sequential affinity purification using anti-FLAG M2 and anti-HA agarose beads and eluted with excessive FLAG and HA peptides. The final eluted materials were resolved by SDS-PAGE and a ∼70-kDa band in PTEN complexes was excised for mass spectrometric analysis.
In vitro protein-protein interaction
Purified His-PTEN and FLAG-PLK1 were incubated in vitro for analysis of their direct interaction. Recombinant proteins FLAG-PLK1, His-PTEN and a His-tagged irrelevant protein, His-Control, were expressed in asynchronous Sf9 cells for 72–96 hours and purified with anti-FLAG M2 beads (Sigma) or Ni-NTA beads (Qiagen). Purified FLAG-PLK1 (0.5 μg) was incubated with His-PTEN (0.5 μg) or His-Control (0.5 μg) for 30 min at room temperature. Protein complexes were then immunoprecipitated with Ni-NTA beads and subjected to SDS-PAGE for detection of FLAG-PLK1 using anti-FLAG antibody.
Dephosphorylation assay in vitro and in vivo
FLAG-PLK1 protein was expressed in Sf9 cells and isolated by immunoprecipitation using anti-FLAG M2 beads. Sf9-expressed His-PTEN protein, as well as 2 mutant forms of PTEN, C124S and G129E, were purified and added to the FLAG immunoprecipitates and incubated at room temperature for 30–60 min. Samples were washed and subjected to Western blot analysis of PLK1 phosphorylation. For in vivo dephosphorylation assay, Pten−/− MEFs were transfected with FLAG-PTEN, FLAG-PTENC124S, FLAG-PTENG129E or an empty control vector. The level of PLK1 phosphorylation was evaluated by immnoblotting with a site-specific anti-phospho-PLK1 (Thr201) antibody.
Abbreviations
| APC | = | anaphase-promoting complex |
| CIN | = | chromosome instability |
| DTB | = | double-thymidine block |
| NudC | = | Nuclear distribution gene C |
| PI3K | = | phosphatidylinositol 3-kinase |
| PIP3 | = | phosphatidylinositol-3,4,5-triphosphate |
| PLK1 | = | polo-like kinase 1 |
| PTEN | = | phosphatase and tensin homolog |
| shRNA | = | short hairpin RNA |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary files
Download PDF (1.1 MB)Funding
This work was supported by NIH grants R01 CA133008 to Y.Y. and R01 GM100478 to W.H.S.
References
- Puc J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L, Mansukhani M, Murty VV, Gaciong Z, Meek SE, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 2005; 7:193-204; PMID:15710331; http://dx.doi.org/10.1016/j.ccr.2005.01.009
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007; 128:157-70; PMID:17218262; http://dx.doi.org/10.1016/j.cell.2006.11.042
- Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O'Neil J, Gutierrez A, Ivanova E, Perna I, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007; 447:966-71; PMID:17515920; http://dx.doi.org/10.1038/nature05886
- Ehlers JP, Worley L, Onken MD, Harbour JW. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res 2008; 14:115-22; PMID:18172260; http://dx.doi.org/10.1158/1078-0432.CCR-07-1825
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A 1999; 96:4240-5; PMID:10200246; http://dx.doi.org/10.1073/pnas.96.8.4240
- Parsons R. Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol 2004; 15:171-6; PMID:15209376; http://dx.doi.org/10.1016/j.semcdb.2003.12.021
- Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol 2009; 4:127-50; PMID:18767981; http://dx.doi.org/10.1146/annurev.pathol.4.110807.092311
- Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, Pandolfi PP. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 2011; 144:187-99; PMID:21241890; http://dx.doi.org/10.1016/j.cell.2010.12.020
- Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013; 341:395-9; PMID:23888040; http://dx.doi.org/10.1126/science.1236188
- Chen ZH, Zhu M, Yang J, Liang H, He J, He S, Wang P, Kang X, McNutt MA, Yin Y, et al. PTEN interacts with histone H1 and controls chromatin condensation. Cell reports 2014; 8:2003-14; PMID:25199838; http://dx.doi.org/10.1016/j.celrep.2014.08.008
- Gong L, Govan JM, Evans EB, Dai H, Wang E, Lee SW, Lin HK, Lazar AJ, Mills GB, Lin SY. Nuclear PTEN tumor-suppressor functions through maintaining heterochromatin structure. Cell Cycle 2015; 14:2323-32; PMID:25946202; http://dx.doi.org/10.1080/15384101.2015.1044174
- He J, Kang X, Yin Y, Chao KS, Shen WH. PTEN regulates DNA replication progression and stalled fork recovery. Nat Commun 2015; 6:7620; PMID:26158445; http://dx.doi.org/10.1038/ncomms8620
- Kang X, Song C, Du X, Zhang C, Liu Y, Liang L, He J, Lamb K, Shen WH, Yin Y. PTEN stabilizes TOP2A and regulates the DNA decatenation. Sci Rep 2015; 5:17873; PMID:26657567; http://dx.doi.org/10.1038/srep17873
- Yu S, Yang F, Shen WH. Genome maintenance in the context of 4D chromatin condensation. Cell Mol Life Sci: CMLS 2016; 73:3137-150; PMID:27098512; http://dx.doi.org/10.1007/s00018-016-2221-2
- Baker SJ. PTEN enters the nuclear age. Cell 2007; 128:25-8; PMID:17218252; http://dx.doi.org/10.1016/j.cell.2006.12.023
- Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene 2008; 27:5443-53; PMID:18794879; http://dx.doi.org/10.1038/onc.2008.241
- Saal LH, Gruvberger-Saal SK, Persson C, Lovgren K, Jumppanen M, Staaf J, Jonsson G, Pires MM, Maurer M, Holm K, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet 2008; 40:102-7; PMID:18066063; http://dx.doi.org/10.1038/ng.2007.39
- Sun Z, Huang C, He J, Lamb KL, Kang X, Gu T, Shen WH, Yin Y. PTEN C-terminal deletion causes genomic instability and tumor development. Cell Rep 2014; 6:844-54; PMID:24561254; http://dx.doi.org/10.1016/j.celrep.2014.01.030
- Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev 2007; 17:157-62; PMID:17324569; http://dx.doi.org/10.1016/j.gde.2007.02.011
- Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 2004; 5:45-54; PMID:14708009; http://dx.doi.org/10.1038/nrm1276
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005; 437:1043-7; PMID:16222300; http://dx.doi.org/10.1038/nature04217
- Storchova Z, Breneman A, Cande J, Dunn J, Burbank K, O'Toole E, Pellman D. Genome-wide genetic analysis of polyploidy in yeast. Nature 2006; 443:541-7; PMID:17024086; http://dx.doi.org/10.1038/nature05178
- Sunkel CE, Glover DM. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci 1988; 89(Pt 1):25-38; PMID:3417791
- Ohkura H, Hagan IM, Glover DM. The conserved Schizosaccharomyces pombe kinase plo1, required to form a bipolar spindle, the actin ring, and septum, can drive septum formation in G1 and G2 cells. Genes Dev 1995; 9:1059-73; PMID:7744248; http://dx.doi.org/10.1101/gad.9.9.1059
- Hartwell LH, Mortimer RK, Culotti J, Culotti M. Genetic control of the cell division cycle in yeast: V. genetic analysis of cdc mutants. Genetics 1973; 74:267-86; PMID:17248617
- Qian YW, Erikson E, Li C, Maller JL. Activated polo-like kinase Plx1 is required at multiple points during mitosis in Xenopus laevis. Mol Cell Biol 1998; 18:4262-71; PMID:9632810; http://dx.doi.org/10.1128/MCB.18.7.4262
- Stegmeier F, Visintin R, Amon A. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell 2002; 108:207-20; PMID:11832211; http://dx.doi.org/10.1016/S0092-8674(02)00618-9
- Yoshida S, Kono K, Lowery DM, Bartolini S, Yaffe MB, Ohya Y, Pellman D. Polo-like kinase Cdc5 controls the local activation of Rho1 to promote cytokinesis. Science 2006; 313:108-11; PMID:16763112; http://dx.doi.org/10.1126/science.1126747
- Petronczki M, Glotzer M, Kraut N, Peters JM. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell 2007; 12:713-25; PMID:17488623; http://dx.doi.org/10.1016/j.devcel.2007.03.013
- Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, Jallepalli PV. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci U S A 2007; 104:4383-8; PMID:17360533; http://dx.doi.org/10.1073/pnas.0701140104
- Brennan IM, Peters U, Kapoor TM, Straight AF. Polo-like kinase controls vertebrate spindle elongation and cytokinesis. PLoS One 2007; 2:e409; PMID:17476331; http://dx.doi.org/10.1371/journal.pone.0000409
- Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene 2005; 24:267-76; PMID:15640842; http://dx.doi.org/10.1038/sj.onc.1208273
- Qian YW, Erikson E, Maller JL. Mitotic effects of a constitutively active mutant of the Xenopus polo-like kinase Plx1. Mol Cell Biol 1999; 19:8625-32; PMID:10567586; http://dx.doi.org/10.1128/MCB.19.12.8625
- Alexandru G, Uhlmann F, Mechtler K, Poupart MA, Nasmyth K. Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast. Cell 2001; 105:459-72; PMID:11371343; http://dx.doi.org/10.1016/S0092-8674(01)00362-2
- Carmena M, Riparbelli MG, Minestrini G, Tavares AM, Adams R, Callaini G, Glover DM. Drosophila polo kinase is required for cytokinesis. J Cell Biol 1998; 143:659-71; PMID:9813088; http://dx.doi.org/10.1083/jcb.143.3.659
- Kneisel L, Strebhardt K, Bernd A, Wolter M, Binder A, Kaufmann R. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutan Pathol 2002; 29:354-8; PMID:12135466; http://dx.doi.org/10.1034/j.1600-0560.2002.290605.x
- Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, Altmannsberger HM, Rubsamen-Waigmann H, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997; 14:543-9; PMID:9053852; http://dx.doi.org/10.1038/sj.onc.1200862
- Takai N, Miyazaki T, Fujisawa K, Nasu K, Hamanaka R, Miyakawa I. Expression of polo-like kinase in ovarian cancer is associated with histological grade and clinical stage. Cancer Lett 2001; 164:41-9; PMID:11166914; http://dx.doi.org/10.1016/S0304-3835(00)00703-5
- Takai N, Miyazaki T, Fujisawa K, Nasu K, Hamanaka R, Miyakawa I. Polo-like kinase (PLK) expression in endometrial carcinoma. Cancer Lett 2001; 169:41-9; PMID:11410324; http://dx.doi.org/10.1016/S0304-3835(01)00522-5
- Knecht R, Elez R, Oechler M, Solbach C, von Ilberg C, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res 1999; 59:2794-7; PMID:10383133
- Kelm O, Wind M, Lehmann WD, Nigg EA. Cell cycle-regulated phosphorylation of the Xenopus polo-like kinase Plx1. J Biol Chem 2002; 277:25247-56; PMID:11994303; http://dx.doi.org/10.1074/jbc.M202855200
- Hamanaka R, Smith MR, O'Connor PM, Maloid S, Mihalic K, Spivak JL, Longo DL, Ferris DK. Polo-like kinase is a cell cycle-regulated kinase activated during mitosis. J Biol Chem 1995; 270:21086-91; PMID:7673138; http://dx.doi.org/10.1074/jbc.270.36.21086
- Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455:119-23; PMID:18615013; http://dx.doi.org/10.1038/nature07185
- Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008; 320:1655-8; PMID:18566290; http://dx.doi.org/10.1126/science.1157425
- Jang YJ, Ma S, Terada Y, Erikson RL. Phosphorylation of threonine 210 and the role of serine 137 in the regulation of mammalian polo-like kinase. J Biol Chem 2002; 277:44115-20; PMID:12207013; http://dx.doi.org/10.1074/jbc.M202172200
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006; 441:475-82; PMID:16598206; http://dx.doi.org/10.1038/nature04703
- Guo W, Lasky JL, Chang CJ, Mosessian S, Lewis X, Xiao Y, Yeh JE, Chen JY, Iruela-Arispe ML, Varella-Garcia M, et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 2008; 453:529-33; PMID:18463637; http://dx.doi.org/10.1038/nature06933
- Guo W, Schubbert S, Chen JY, Valamehr B, Mosessian S, Shi H, Dang NH, Garcia C, Theodoro MF, Varella-Garcia M, et al. Suppression of leukemia development caused by PTEN loss. Proc Natl Acad Sci U S A 2011; 108:1409-14; PMID:21212363; http://dx.doi.org/10.1073/pnas.1006937108
- Lee KS, Erikson RL. Plk is a functional homolog of Saccharomyces cerevisiae Cdc5, and elevated Plk activity induces multiple septation structures. Mol Cell Biol 1997; 17:3408-17; PMID:9154840; http://dx.doi.org/10.1128/MCB.17.6.3408
- Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet 2010; 42:454-8; PMID:20400965; http://dx.doi.org/10.1038/ng.556
- Shen-Li H, Koujak S, Szablocs M, Parsons R. Reduction of Pten dose leads to neoplastic development in multiple organs of Pten (shRNA) mice. Cancer Biol Ther 2010; 10:1194-200; PMID:20980828; http://dx.doi.org/10.4161/cbt.10.11.13814
- Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol 2003; 13:478-83; PMID:12946627; http://dx.doi.org/10.1016/S0962-8924(03)00175-2
- Tamguney T, Stokoe D. New insights into PTEN. J Cell Sci 2007; 120:4071-9; PMID:18032782; http://dx.doi.org/10.1242/jcs.015230
- Zhou T, Aumais JP, Liu X, Yu-Lee LY, Erikson RL. A role for Plk1 phosphorylation of NudC in cytokinesis. Dev Cell 2003; 5:127-38; PMID:12852857; http://dx.doi.org/10.1016/S1534-5807(03)00186-2
- Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res 2011; 71:2821-5; PMID:21385900; http://dx.doi.org/10.1158/0008-5472.CAN-10-3399
- Tibarewal P, Zilidis G, Spinelli L, Schurch N, Maccario H, Gray A, Perera NM, Davidson L, Barton GJ, Leslie NR. PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci Signaling 2012; 5:ra18; PMID:22375056; http://dx.doi.org/10.1126/scisignal.2002138
- Nelen MR, van Staveren WC, Peeters EA, Hassel MB, Gorlin RJ, Hamm H, Lindboe CF, Fryns JP, Sijmons RH, Woods DG, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet 1997; 6:1383-7; PMID:9259288; http://dx.doi.org/10.1093/hmg/6.8.1383
- Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997; 16:64-7; PMID:9140396; http://dx.doi.org/10.1038/ng0597-64
- Holt LJ. Regulatory modules: Coupling protein stability to phopshoregulation during cell division. FEBS Lett 2012; 586:2773-7; PMID:22664379; http://dx.doi.org/10.1016/j.febslet.2012.05.045
- Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 2007; 17:304-15; PMID:17291761; http://dx.doi.org/10.1016/j.cub.2006.12.046
- Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol 2007; 17:316-22; PMID:17291758; http://dx.doi.org/10.1016/j.cub.2006.12.037
- Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF, Cheng L, Ahmad N, Liu X. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem 2011; 286:35795-800; PMID:21890624; http://dx.doi.org/10.1074/jbc.C111.269050
- Li Z, Li J, Bi P, Lu Y, Burcham G, Elzey BD, Ratliff T, Konieczny SF, Ahmad N, Kuang S, et al. Plk1 phosphorylation of PTEN causes a tumor-promoting metabolic state. Mol Cell Biol 2014; 34:3642-61; PMID:25047839; http://dx.doi.org/10.1128/MCB.00814-14
- Choi BH, Pagano M, Dai W. Plk1 protein phosphorylates phosphatase and tensin homolog (PTEN) and regulates its mitotic activity during the cell cycle. J Biol Chem 2014; 289:14066-74; PMID:24706748; http://dx.doi.org/10.1074/jbc.M114.558155
- Sagona AP, Nezis IP, Pedersen NM, Liestol K, Poulton J, Rusten TE, Skotheim RI, Raiborg C, Stenmark H. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol 2010; 12:362-71; PMID:20208530; http://dx.doi.org/10.1038/ncb2036
- Bolanos-Garcia VM. Assessment of the mitotic spindle assembly checkpoint (SAC) as the target of anticancer therapies. Curr Cancer Drug Targets 2009; 9:131-41; PMID:19275754; http://dx.doi.org/10.2174/156800909787580980
- Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell 2009; 16:347-58; PMID:19800579; http://dx.doi.org/10.1016/j.ccr.2009.08.020
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell 2008; 133:612-26; PMID:18485870; http://dx.doi.org/10.1016/j.cell.2008.03.025