
Aerobic glycolysis, or the Warburg effect, has long been implicated in cancer cell metabolism, growth, and survival.Citation1 Consequently, therapeutic targeting of glycolysis in cancer has been considered an attractive proposition. Unfortunately, though, this treatment strategy has not yet been realized in the clinic. Cancer cells can leverage their substantial metabolic plasticity to circumvent the potentially deleterious effects of inhibiting any single metabolic pathway, and, a deep understanding of the metabolic plasticity that underlies the adaptive response to glycolytic inhibition is lacking. We explored this metabolic plasticity in cancer cell line models.Citation2
We began by screening a panel of tumor-derived cell lines for sensitivity to the glycolysis inhibitor 2-deoxy-D-glucose (2DG). Cells that were highly sensitive to 2DG were denoted as glycolysis-dependent (GD) and those that were less sensitive were defined as glycolysis-independent (GI). To model the acquisition of glycolysis independence and metabolic plasticity in 2DG-sensitive GD cells, we continuously propagated such cells in increasing 2DG concentrations until they exhibited at least 10-fold reduced 2DG sensitivity. We found that GI cells (those that started out that way or were generated from GD cells under selection), but not GD cells, compensate for reduced glycolysis by up-regulating oxidative phosphorylation (OXPHOS) both at basal and glycolysis-inhibited conditions (). We observed that glutamine-carbon supports and sustains the increased OXPHOS in GI cells. Glutamine-carbon provides acetyl coenzyme (ACoA) for OXPHOS in the TCA cycle.Citation3,4 However, curiously, glutamine was not the source for ACoA in GI cells. Strikingly, GI cells, but not GD cells, were able to circumvent the 2DG-induced glycolysis block via the pentose phosphate pathway (PPP) to generate not only ACoA but also lipids and nucleotides from glucose itself. This observation has critical implications for GI cell anabolism, growth and proliferation and implicates glucose-carbon as a source of cellular ATP and macromolecular necessities.
Figure 1. mTORC1 re-wires GI cell metabolism upon glycolysis inhibition to promote growth and proliferation. Green arrows indicate either increased mTORC1-mediated pathway activity or higher levels of the indicated enzymes or metabolites. Enzymes are italicized. G6P, glucose 6-phosphate; F6P, fructose 6-phosphate; F1,6P, fructose 1,6-diphosphate; G3P, glyceraldehyde 3-phosphate; 3PG, 3-phosphoglycerate; 2PG, 2-phosphoglycerate; PEP, phosphoenol pyruvate; PYR, pyruvate; LAC, lactate; ACoA, acetyl CoA; SER, serine; GLY, glycine; 6PGL, 6-phosphogluconolactone; 6PG, 6-phosphogluconate; Ru5P, X5P, R5P, ribulose-, xylulose-, and ribose- 5 phosphates; S7P, sedoheptulose 7-phosphate; GPI, glucose-6-phosphate isomerase; G6PD, glucose-6-phosphate dehydrogenase; CAD, CAD enzyme.
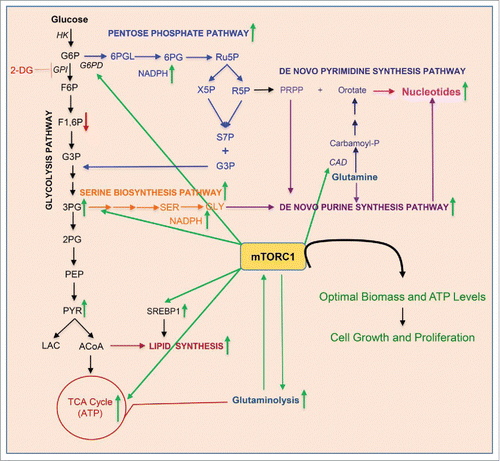
Next, we probed the mechanistic basis of the metabolic reprogramming observed in GI cells. The mTOR pathway is a key regulator of cellular energy and biomass production and is inhibited by 2DG.Citation5,6 Remarkably, 2DG inhibited the S6 kinase (S6K) axis of mTORC1 only in GD cells but not in GI cells. In addition, the mTORC1/S6K axis is sustained only in GI cells that are resistant to other glycolysis inhibitors, oxamate and GNE-140, which inhibit lactate dehydrogenase A (LDHA). Consistent with our observation that GI cells circumvent the 2DG-induced glycolysis block via PPP, expression of the rate-limiting PPP enzyme, glucose-6-phosphate dehydrogenase (G6PD) was elevated by 3-fold in GI but not in GD cells. Strikingly, this upregulation is dependent upon mTORC1. Moreover, in GI cells, but not in GD cells, mTORC1 sustains the activation status of the lipidogenic transcription factor SREBP1 and the enzyme CAD, a key regulator of de novo pyrimidine synthesis. Therefore, by orchestrating the metabolic re-wiring of GI cells, mTORC1 provides for their energy and biomass demands upon glycolytic stress.
As proof of principle for possible therapeutic utility, we tested the consequences of dual inhibition of glycolysis and mTORC1 on GI cell metabolism and viability. Whereas the pharmacological inhibition of glycolysis or mTORC1/S6K alone showed no deleterious effects on GI cell viability, their combined inhibition synergized to significantly reduce nucleotide, TCA cycle metabolite levels, cell viability and clonogenic growth. We were able to recapitulate the synergy using an orthogonal approach by genetically knocking down glucose-6 phosphate isomerase (GPI), the enzymatic target of 2DG. Remarkably, only the combination of GPI knockdown and everolimus, a clinical mTORC1 inhibitor, significantly reduced GI cell xenograft tumor growth in vivo.
This analysis revealed for the first time a remarkable degree of coordinated regulation of multiple cellular metabolic nodes by mTORC1 in the context of acquisition and maintenance of glycolysis independence. For example, mTORC1, by upregulating PPP enzymes, not only circumvents the 2DG-induced glycolysis block, but, in the process, also significantly increases NADPH levels in GI cells. NADPH provides critical reducing power to negate enhanced ROS (reactive oxygen species) levels that would be expected due to higher OXPHOS levels in GI cells, which in turn, were upregulated by mTORC1. Furthermore, mTORC1-enhanced lipid synthesis, a highly NADPH-demanding pathway, would benefit from increased NADPH levels in GI cells.
Although we provided some insight into the probable mechanism as to how the mTORC1/S6K axis is sustained in GI cells, it certainly requires further investigation. In GD cells, but not in GI cells, multiple stress signaling pathways converge on mTORC1 and negatively regulate it upon glycolysis inhibition. However, the mechanism by which GI cells avoid glycolytic stress itself still needs to be addressed. Additionally, our data suggest significant cross-talk and interdependency between glutamine metabolism and mTORC1 signaling. However, it remains unclear whether glutaminolysis initially drives and sustains mTORC1 signaling or whether the converse is true during the acquisition of glycolysis independency.
Our findings suggest that mTORC1 function allows cancer cells to circumvent the deleterious effects of the inhibition of more than one glycolysis-associated enzyme. This implies a significant degree of similarity in the mechanisms underlying escape from glycolysis dependency, which reinforces the rationale for testing the combination of glycolysis and mTORC1 inhibitors to realize the potential therapeutic utility of targeting the Warburg effect.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324(5930):1029-1033; PMID:19460998; http://dx.doi.org/10.1126/science.1160809
- Pusapati RV, Daemen A, Wilson C, Sandoval W, Gao M, Haley B, Baudy AR, Hatzivassiliou G, Evangelista M, Settleman J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016; 29:548-562; PMID:27052953; http://dx.doi.org/10.1016/j.ccell.2016.02.018
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012; 481:385-388; PMID:22101431; http://dx.doi.org/10.1038/nature10642
- Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell 2014; 56:414-424; PMID: 25458842; http://dx.doi.org/10.1016/j.molcel.2014.09.025
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-293; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science 2001; 294:1102-1105; PMID:11691993; http://dx.doi.org/10.1126/science.1063518