
New causal treatment options against chronic kidney diseases (CKD) represent a major unmet medical need. Glomerular diseases account for nearly 85% of endstage renal disease cases in the USA and Western Europe and progressive podocyte loss appears to be a common underlying cause of glomerulosclerosis.Citation1 Despite considerable progress in deciphering the molecular pathomechanisms leading to glomerulosclerosis causal treatment options slowing down or even halting the progression of glomerular diseases remain very limited.
In their recent paper published in Cell Cycle Hagen and colleagues can successfully recapitulate the formation of bi- and multinucleated podocytes in an in vitro model of differentiated postmitotic mouse podocytes.Citation2 These peculiar polynucleated podocytes have been documented in diverse glomerular diseases ranging from Focal-Segmental-Glomerulo-Sclerosis (FSGS), Lupus Nephritis, IgA Nephropathy to Diabetic and Hypertensive Nephropathy and are often accompanied by cell hypertrophy.Citation3 So far it was unclear whether this aberrant and incomplete mitosis is part of a glomerular repair mechanism by which podocytes enlarge to cover denuded glomerular basement membrane areas or whether this reentry into cell cycle rather increases the susceptibility and subsequent loss of the affected cell.Citation3 Hagen and colleagues now demonstrate that upon stimulation with growth factors such as basic FGF or TGFβ terminally differentiated podocytes grown under non-permissive conditions started reentering the cell-cycle as evidenced by re-expression of the proliferation markers KI67 and phosphorylated-histone H3. In a considerable proportion of cells this reentry into cell-cycle led to an incomplete mitosis and resulted in polynucleated cells. This is in agreement with previous findings demonstrating that podocytes cannot assemble an efficient mitotic spindle, which is essential for cytokinesis and the completion of mitosis.Citation4
The polynucleation did not decrease cellular viability per se, but enhanced the susceptibility to podocyte stress models such as Puromycin Aminonucleoside (PAN). This intriguing finding helps to draw a clearer picture of glomerular pathomechanisms: Glomerular injury leads to increased secretion of growth factors such as basic FGF or TGFß. This stimulus, which might primarily be aimed at inducing a compensatory cell hypertrophy, also induces re-expression of Cyclin A and CDK2, subsequently driving some of these cells into an aberrant G2/M phase. This culminates in an aborted mitosis resulting in polynucleated podocytes or eventual direct cell death (catastrophic mitosis).Citation3,5 While the impact, timing and function of cellular podocyte hypertrophy have recently been intensively studied, the role of multi-nucleation as a “by-product” of growth-promoting pathways remained elusive.Citation6 Here the authors highlight that podocyte multinucleation per se leave podocytes in a vulnerable state to ”second hits” which ultimately drive podocyte loss.
As alluded by the authors this newly established in vitro model can be used to screen for substances that either prevent entry into aberrant mitosis or ameliorate subsequent “second hits” on polynucleated podocytes. While the in vitro evidence for this concept is convincingly demonstrated by Hagen and colleagues, further research is required to translate these findings into an in vivo model to prove the sequence of glomerular events and to ultimately confirm new treatment options identified in the in vitro screens. In addition, more pathomechanistically relevant stimuli such as hypertension, diabetes or immune mediated injury should be tested in vivo. It might turn out that the balance of podocyte hypertrophy needed to cover the GBM at sites of podocyte loss and the “prize” of a decreased stress response, will be very different in an in vivo situation compared to cultured podocytes without the demand to shield the renal filter.
While in the authors’ in vitro system podocytes might indeed undergo apoptosis, the occurrence of this special form of cell death is highly debated in the in vivo situation.Citation1,3,7 In fact, evidence for podocyte apoptosis in vivo is extremely scarce if not completely absent, despite reports of expression of bona fide apoptosis related genes and proteins in podocytes. It seems more likely that severely injured podocytes detach from the glomerular basement membrane and are lost into the urine.Citation7
Together, the report of Hagen and colleagues sheds new light on the intricate balance of compensatory actions and stress response capabilities of podocytes.
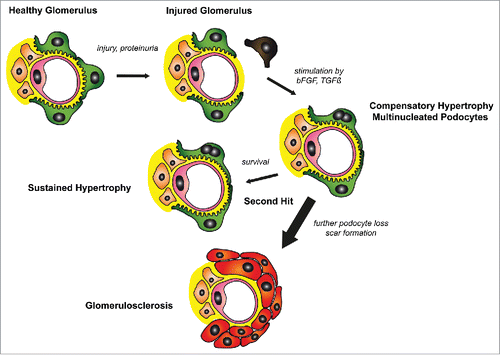
Figure 1. A potential in vivo translation of the findings by Hagen et al.Citation2 : Glomerular injury causes loss of podocytes. Release of growth factors such as bFGF or TGFß induces compensatory hypertrophy of remaining podocytes. In a percentage of podocytes these growth factors also trigger initiation of the cell cycle leading to aberrant mitosis and multinucleated cells. Multinucleated podocytes are able to shield the glomerular wall, yet are more susceptible to a second hit. Further loss of podocytes will ultimately result in glomerulosclerosis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Nicola Wanner for help with art work.
Funding
The laboratory of the authors is generously supported by the German Research Foundation (DFG): CRC 1140 (to FG and TBH), CRC 992 (to TBH) and Heisenberg program (to TBH) and HU 1016/8-1; by the European Research Council-ERC grant 616891 (to TBH); by the BMBF-Joint transnational Grant STOP-FSGS 01GM1518C (to TBH) and by the Excellence Initiative of the German Federal and State Governments (EXC294, BIOSS II to TBH).
References
- Tharaux PL, Huber TB. How many ways can a podocyte die? Semin Nephrol 2012; 32:394–404; PMID:22958494; http://dx.doi.org/10.1016/j.semnephrol.2012.06.011
- Hagen M, Pfister E, Kosel A, Shankland S, Pippin J, Amann K, Daniel C. Cell cycle re-entry sensitizes podocytes to injury induced death. Cell Cycle 2016; 15(14):1929-37; PMID: 27232327; http://dx.doi.org/10.1080/15384101.2016.1191710
- Liapis H, Romagnani P, Anders HJ. New insights into the pathology of podocyte loss: mitotic catastrophe. Am J Pathol 2013; 183:1364–74; PMID:24007883; http://dx.doi.org/10.1016/j.ajpath.2013.06.033
- Lasagni L, Ballerini L, Angelotti ML, Parente E, Sagrinati C, Mazzinghi B, Peired A, Ronconi E, Becherucci F, Bani D, et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells 2010; 28:1674–85; PMID:20680961; http://dx.doi.org/10.1002/stem.492
- Brinkkoetter PT, Olivier P, Wu JS, Henderson S, Krofft RD, Pippin JW, Hockenbery D, Roberts JM, Shankland SJ. Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells. J Clin Investig 2009; 119:3089–101; PMID:19729834; http://dx.doi.org/10.1172/JCI37978
- Grahammer F, Wanner N, Huber TB. mTOR controls kidney epithelia in health and disease. Nephrol Dial Transplant 2014; 29(Suppl 1):i9-i18; http://dx.doi.org/10.1093/ndt/gft491
- Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol 2015; 26:258–69; PMID:25060060; http://dx.doi.org/10.1681/ASN.2014030278