
ASBTRACT
Increased rates of cholesterol and lipid synthesis have long been recognized as important aspects of the metabolic rewiring that occurs during cancerous transformation. Many genes encoding enzymes involved in cholesterol and fatty acid biogenesis are transcriptional targets of the sterol regulatory element-binding proteins (SREBPs). The SREBPs act as a hub for metabolic and proliferation-related signals; their activity is the focus of a tug-of-war between tumor suppressors, who generally inhibit SREBP function, and oncogenes, who often promote, and rely on, SREBP activity. The Hippo pathway plays a central role in coordinating cell proliferation and organ size, whereas p53 is a crucial tumor suppressor that maintains metabolic homeostasis and orchestrates cellular stress responses. Together, the Hippo and p53 signaling pathways cooperate on multiple levels to fine-tune SREPB activity and regulate cholesterol/lipid levels. Cholesterol biosynthesis inhibitors such as statins are appealing conceptually, but have yet to show an indisputable effect on cancer development. Fortunately, the complex regulation surrounding the Hippo-p53-SREBP network potentially provides a broad interface for additional novel cancer-targeting interventions.
Introduction
Cholesterol is a unique lipid, essential for membrane biogenesis, cell proliferation and cell differentiation.Citation1 It is the precursor of steroid hormones, bile acids and vitamin D. Cholesterol is available through diet, but is also synthesized by the liver, the small and large intestines, and additional tissues. It is distributed throughout the body via low-density lipoprotein (LDL) and high-density lipoprotein (HDL) transporters. Mammalian cells synthesize cholesterol through a multi-step enzymatic process, generating numerous metabolites that mediate physiological, developmental and tumorigenic processes.Citation1
Excess cholesterol was shown to promote mammary tumor growth and invasiveness in several mouse transgenic models.Citation2-4 In humans, hypercholesterolemia is an independent risk factor for breast cancerCitation5-7 and for decreased response of tumors to endocrine therapies.Citation8 This is partially due to the fact that some cholesterol metabolites (such as 27-hydroxycholesterol), although different in structure, can have similar modes of action as estrogen, increasing the proliferation of estrogen receptor-positive breast cancer cells.Citation9-11 Of note, other cholesterol-associated metabolites have been shown to inhibit tumorigenesis.Citation12
Cellular cholesterol levels are intimately connected to the SREBP-mevalonate pathway.Citation13 The mammalian genome contains 2 SREBP genes: SREBP1 and SREBP2, which encode related sequence-specific transcription factors. Together, the SREBP proteins directly activate a multitude of genes dedicated to the synthesis and uptake of cholesterol, fatty acids, triglycerides and phospholipids.Citation13,14 SREBP1 mainly regulates lipid biogenesis, whereas genes involved in cholesterol synthesis are predominantly transactivated by SREBP2.Citation15-17 Two isoforms of SREBP1, SREBP1a and SREBP1c, are generated through the use of alternative promoters; SREBP1c (with the shorter transactivation domain and thus weaker transcriptional activity) is predominantly expressed in the liver, whereas in other organs SREBP1a is more prevalent.
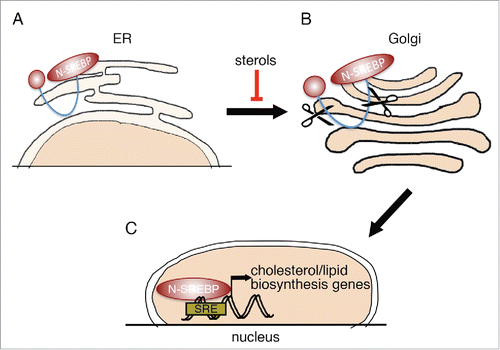
All three proteins are synthesized as inactive precursors (P-SREBP), bound to the endoplasmic reticulum (ER)Citation18 (). When cells become depleted of cholesterol, P-SREBP proteins are escorted to the Golgi apparatus (). In the Golgi, P-SREBP is consecutively cleaved by 2 distinct proteases, so as to release the N-terminal, transcriptionally active domain (N-SREBP, ). N-SREBP enters the nucleus and promotes a lipogenic/cholesterol synthesis program by binding to its consensus sites.Citation14 When cholesterol content in the cell rises, P-SREBPs are retained on the ER membrane, turning off the transcription of target genes.Citation13 In this way, SREBPs both affect and are affected by cellular cholesterol levels.
Figure 1. SREBP processing. (A) SREBP transcription factors are synthesized as inactive precursors (P-SREBP), retained at the membrane of the ER. When intracellular sterol levels become low, P-SREBP is released from the ER membrane and is translocated to the Golgi apparatus. (B) SREBP in the Golgi apparatus undergoes 2 sequential proteolytic cleavage events. (C) The N-terminal part of SREBP (N-SREBP), containing a transcriptional activation domain and a DNA binding domain, is translocated to the nucleus, where it can bind sterol response elements (SREs) and regulate the expression of associated target genes.
LATS2 and p53 cooperate to restrain cellular SREBP activity
Not surprisingly, the SREBPs act as a hub for metabolic and proliferation-related signals and are highly regulated by cancer-associated pathways. Together, they constitute a higher order network that forms a molecular base framework for determining cellular cholesterol levels in health and disease, including cancer. As discussed below, tumor suppressors often inhibit SREBP activity, whereas oncogenes augment and/or are dependent on SREBP activity. This represents a high-stakes battle for metabolic control, which is inevitably corrupted when cells undergo neoplastic transformation.
The Hippo signaling pathway is a critical regulator of cell proliferation and differentiation. Central to this pathway is a core kinase cascade of the tumor suppressors MST1/2, LATS1/2 and the adaptor proteins SAV1 and MOB1/2.Citation19 sThese proteins form a conserved kinase cassette (“Hippo”) that limits tissue growth and progenitor cell proliferation, typically, by phosphorylating and inactivating the transcriptional co-activators YAP and TAZ. Hippo-phosphorylated YAP/TAZ are sequestered in the cytoplasm and are primed for rapid proteasomal degradation, thereby repressing their transcriptional activities and their positive effects on tissue growth and proliferation.Citation20-22 In normal tissues, integrity of the Hippo pathway keeps cells in check and prevents their uncontrolled proliferation. Conversely, dysfunction of the Hippo pathway can lead to constitutive activation of YAP/TAZ, and is associated with many types of cancer.Citation23
p53 is a major tumor suppressor, mutationally inactivated in about half of all cases of human cancer.Citation24,25 In view of its central role in ensuring genome integrity and elimination of cells harboring defective genomes, p53 has been dubbed “guardian of the genome.”Citation26 Several years ago, LATS2 – a key component of the core Hippo pathway- was found to contribute to stabilization and activation of p53 in response to mitotic machinery damage and oncogene activation,Citation27 implicating it as a member of the extended network that relays to p53 signals from several types of genome-endangering stress. Recently, LATS2, together with its paralog LATS1, was shown to contribute to the canonical tumor suppressive features of p53 also under basal conditions, in the absence of pronounced genotoxic stress.Citation28 Furthermore, p53 binds and transactivates the LATS2 promoter, thus defining a positive feedback loop.Citation27,29,30 (). This LATS2-p53 axis intimately couples two important tumor suppressor pathways, p53 and Hippo, not only in response to genotoxic insults but also in physiological processes such as induced cell differentiation.Citation31
Figure 2. LATS2 p53 and cholesterol crosstalk. (A) LATS2 and p53 positively regulate each other, to inhibit cholesterol synthesis. Excessive levels of cholesterol activate the LATS2-p53 axis. Green arrows depict positive regulation and red lines denote negative regulation. (B) Proposed bistability governing cholesterol level-dependent engagement of LATS2-p53. The response to increasing input (upward arrowheads) differs from the response to decreasing input (downward arrowheads). LATS2-p53 activation is triggered above a threshold of cholesterol levels, to reach state “B.” Once the feedback loop is active, it sustains its activity so that the inactivation threshold is shifted to lower cholesterol levels, to reach state “A.” In this way, the feedback loop provides resistance to fluctuations between the “A” and “B” states.
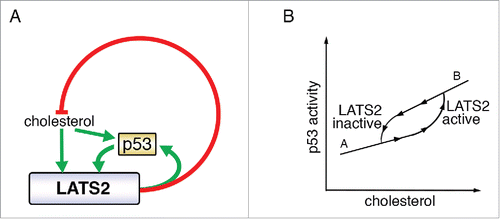
Recently, a new metabolic role of LATS2 was unveiled. Specifically, in both cultured liver-derived human cells and in vivo mouse liver tissue, LATS2 was shown to bind the ER-tethered precursors (P-SREBP) of SREBP1 and SREBP2, inhibiting their processing and quenching the subsequent transcriptional activity of the cleaved, nuclear SREBPsCitation32 (). In cultured liver-derived cells, LATS2 silencing results in constitutive activation of SREBPs and enhancement of their transcriptional signature. Mice harboring liver-specific conditional Lats2 knockout and maintained on normal chow diet accumulate excessive hepatic cholesterol, and spontaneously develop fatty liver disease. When challenged with excess dietary cholesterol, which normally induces a marked p53 response in conjunction with augmented hepatocyte apoptosis, these mice fail to activate p53, and manifest more severe liver dysfunction. Hepatic LATS2 is also important for recovery from high cholesterol-imposed liver damage when mice are returned to regular diet. Consequently, mouse livers lacking Lats2 develop premalignant-like pathology, including ductal reactions and hyperproliferation of oval cells.Citation32 In line with these observations, mice constitutively expressing a SREBP1 transgene have high levels of hepatic p53.Citation33 In parallel, p53 tunes down cellular SREBP activity by repressing the expression of SREBP mRNA.Citation34 Together, these observations underscore the protective homeostatic role of the LATS2-p53 axis, in the liver and possibly also in other cell and tissue types.
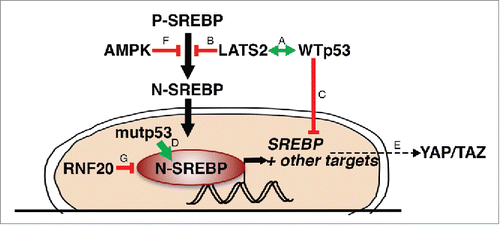
Figure 3. A partial picture of) the SREBP regulatory network. The LATS2-WTp53 axis (A) inhibits processing of P-SREBP (B) and transcription of SREBP mRNA (C). Mutant p53 (mutp53) binds N-SREBP and augments its transcriptional activity (D). YAP/2AZ are dependent on the mevalonate pathway for their activity (E). AMPK inhibits processing of P-SREBP (F). RNF20 ubiquitinates SREBP and drives its degradation (G).
The LATS2-p53-cholesterol feedback loop () conceivably may have evolved to maintain cholesterol homeostasis in healthy individuals. Typically, humans are exposed to fluctuating levels of extracellular cholesterol throughout the day, depending on food intake, and experience a prolonged dip in dietary cholesterol during the night. In this context, it may be beneficial to avoid “wasteful” activation of SREBP (and cholesterol biosynthesis) intermittently during the day, while ensuring sufficient cholesterol synthesis during more extended periods of fasting (night). Indeed, lipid and cholesterol metabolism have been shown to exhibit circadian rhythms.Citation35 One way to achieve 2 alternate behaviors (diurnal low cholesterol = SREBP “off;” nocturnal low cholesterol = SREBP “on”) in humans, in an all-or-none fashion, is by generating a bistable system.Citation36,37 With this type of model in mind, we might envisage the following scenario: low cholesterol levels would not engage the LATS2-p53 feedback loop, so that SREBP could function uninhibited (). During the day, as dietary cholesterol levels increase, the LATS2-p53 axis may become activated, and inhibit SREBP to curb excessive de novo cholesterol biosynthesis (). Once LATS2-p53 are high, their mutual reinforcement through the positive feedback loop would ensure that they remain engaged even at lower concentrations of cholesterol, within the range encountered during the day, at least for several hours. Inactivation of the LATS2-p53 axis might then occur only during nocturnal fasting, when cholesterol levels go beyond the set threshold, thereby facilitating SREBP reactivation (). This model predicts that inactivation of LATS2, as occurs in many cancers,Citation38 would be sufficient to drive excess accumulation of cholesterol () and result in cells trapped in state “A,” despite relatively high levels of cholesterol (). In nocturnal rodents, such as mice, which feed primarily during the night, the 2 phases of this scenario are expected to be reversed relative to humans. Although presently still largely speculative, the predictions of this model are readily amenable to experimental testing.
Networking with SREBP
Important processes in the cell usually are tightly regulated on multiple levels. Moreover, separate and “linear” pathways often intertwine to form integrated and coordinated signaling networks. Therefore, it is not surprising that the Hippo and p53 pathways cross-talk with cholesterol metabolism by additional mechanisms beyond the LATS2-p53 axis described above.
As already noted, p53 transcriptionally represses the expression of SREBP1c as well as of 2 of its lipogenic target genes, fatty acid synthase (FASN) and ATP citrate lyase (ACLY)Citation34 (). Moreover, under glucose starvation p53 induces LIPIN1, a key modulator of SREBP.Citation39,40 Importantly, many of those transcriptional effects are exerted by p53 also under basal conditions, in the absence of notable stress, thereby enabling p53 to fine-tune the lipid metabolic landscape of pertinent cells and tissues.
Gain-of-function mutant forms of p53 (mutp53) occur in approximately 50% of all cancers.Citation41 Interestingly, mutp53 has been shown to bind SREBP1 and SREBP2 and increase the transcription of their target genesCitation42 (). In cells harboring mutp53, upregulation of the mevalonate pathway through this mechanism is sufficient to disrupt cell morphology and drive malignant phenotypes such as invasion.Citation42 Thus, wild type (WT) and mutp53 enact a yin-yang duality of opposite functions, WT p53 inhibiting and mutp53 augmenting SREBP activity. This also suggests that restriction of cholesterol production is central to the tumor suppressive role of WT p53.
As mentioned above, YAP/TAZ are the downstream effectors of the Hippo pathway. Characteristically, YAP/2AZ protein levels and transcriptional activity are inhibited by the LATS1/2 kinases.Citation21,43 Recent studiesCitation44-46 have revealed that the cancer-promoting features of YAP/TAZ are dependent on cholesterol and SREBP-mevalonate pathway activity (). Mechanistically, sustained YAP/TAZ nuclear activity, in association with reduced YAP/TAZ inhibitory phosphorylation, is linked to geranylgeranylation of Rho small GTPasesCitation44 and F-actin cytoskeleton integrity,Citation45 both of which are regulated by SREBP. In turn, YAP itself can facilitate transcription of several genes involved in cholesterol metabolism.Citation45 In fact, mutp53 and YAP physically interact and cooperatively drive transcription. Intriguingly, inhibition of the mevalonate pathway, by treatment with statins, was shown to reduce the transcriptional effects of mutp53 in a YAP-dependent manner.Citation47 Together, these observations suggest that the mevalonate pathway is crucial both as an upstream regulator and as a downstream effector of mutp53 and YAP oncogenic functions.
Of note, in the above studies, LATS1/2 appear to be dispensable for YAP/TAZ phosphorylation in response to statin treatment;Citation44,45 this is inconsistent with the notion that statins operate through a simple linear Hippo pathway. Notably, LATS1/2-independent YAP activity has been observed also in other contexts,Citation48 implying that other kinases are also capable of targeting and inhibiting YAP/TAZ. One interesting candidate might be AMP-activated protein kinase (AMPK),Citation49-51 a master regulator of metabolism, and itself an inhibitor of SREBP processingCitation52 (). It will be interesting to unravel the molecular events that override canonical Hippo signaling and bypass the negative regulation of YAP/TAZ by LATS1/2.
Similarly, some tumor suppressive functions of Hippo pathway components appear to be YAP-independent.Citation53 Indeed, LATS2 has a growing spectrum of functions, including maintenance of genome stability, induction of apoptosis, cell cycle and tetraploidy checkpoint control, inhibition of cell migration and regulation of stem cell differentiation, which extend beyond YAP/TAZ regulation.Citation27,28,31,38,54-63 Likewise, the role of LATS2 in inhibiting SREBP1/2 is independent of YAP.Citation32 Thus, LATS2 appears to engage a multi-pronged strategy to inhibit SREBP activity, by (1) directly binding to and inhibiting processing of cytoplasmic P-SREBP,Citation32 (2) upregulating p53 protein levels and thereby presumably augmenting p53's SREBP-inhibitory effect, and (3) conceivably also by acting, together with LATS1, via the canonical Hippo pathway, to inhibit YAP nuclear accumulation, thereby indirectly dampening YAP-dependent SREBP output.Citation45
Undeniably, SREBP activity is regulated by a slew of factors beyond those mentioned above. For instance, mature N-SREBPs are unstable proteins, highly regulated by ubiquitination and proteasomal degradation.Citation64,65 The RNF20 protein is an E3 ligase that primarily drives histone H2B monoubiquitylation to regulate gene expression and cancer-related features.Citation66-68 Yet, a recent reportCitation69 has associated RNF20 also with ubiquitylation and enhanced proteasomal degradation of SREBP (). Interestingly, depletion of RNF20 decreases the transcription of p53,Citation66 implying yet another potential mechanism for coordination between tumor suppression and cholesterol synthesis.
Thus, as depicted in , the Hippo, RNF20 and p53 pathways impact cellular SREBP activity at different regulatory steps: expression of SREBP mRNA, processing of SREBP proteins, SREBP transcriptional activity, and protein degradation. Consequently, releasing SREBP from its inhibitory constraints to turn “on” cholesterol biosynthesis necessitates disengagement of more than one “brake.” Besides underscoring the importance of restraining cellular cholesterol levels, what might be the logic of this multi-layered system of SREBP regulation? In seeking the answer, 3 important parameters can be considered: kinetics (response time), optimization (energy expenditure) and noise (expression variation). Since protein synthesis is energetically costly for the cell, the “cheapest” way to turn on/off a signal is by activating/inhibiting transcription.Citation70 Transcriptional control is p53's forte ,Citation71 hence p53 represses SREBP, at least in part, at the transcriptional level ().Citation34 However, relying exclusively on transcription may be detrimental for essential functions such as cholesterol maintenance. Moreover, transcriptional bursts are the source of high expression variability (noise)Citation72; unpredictable fluctuations in the rate of cholesterol biosynthesis, caused by excess transcriptional noise, may be hazardous for cells with tight energy constraints.
Regulation of protein processing and compartmentalization often evolve to solve the challenges of toxic pathway intermediates, competing enzymatic reactions or slow turnover rates.Citation73 In general, post-translational control offers a rapid response rate. One effective post-translational mechanism is proteolytic activation of a key regulatory protein, as exemplified by SREBP. Yet, since proteolysis is a thermodynamically favorable and irreversible reaction, prevention of uncontrolled processing through temporal and compartmentalized occlusion, e.g. to the ER in the case of P-SREBP, is of advantage to the robustness of the pathway. On the other hand, post-translational covalent modifications, particularly phosphorylation, are rapidly reversible, and allow existing proteins to toggle between activated/inactivated states without the need for de novo protein synthesis or proteolytic processing.Citation74 However, both modes of post-translational regulation come at a high energetic cost of producing proteins that might potentially remain in their “wasteful” inactive state, if an appropriate activating trigger is not encountered by the cell.
Coordination of transcription with the translocation of P-SREBP from the ER and the processing of Golgi-tethered P-SREBP, might glean the combined benefits of both regulatory levels. Turning off SREBP transcription and processing might constitute a dual fail-safe “brake” against undue cholesterol synthesis. On the other hand, modulation of the extent of post-translational inhibition might be a method to buffer a potentially noisy transcriptional signal and might avoid the buildup of potentially bulky intermediates embedded in the ER membrane. Protein ubiquitination and proteasomal degradation represent a more extreme form of post-translational control; degradation is indisputably an irreversible process. RNF20-mediated ubiquitylation and subsequent proteasomal degradation of SREBPCitation69 () might represent a last resort to restrict SREBP function.
Swapping protein binding partners is another flexible, dynamic strategy to control “quick-response” signaling pathways.Citation75 Protein interaction partners have the advantage of increased functional flexibility, being able to change over time and space to adapt to different conditions. However, cancer cells are infamously adept at hijacking cellular signaling mechanisms for their own sinister cause.Citation76 Thus, the reversibility and transience of SREBP associations with partner proteins might also make them particularly susceptible to hijacking by pro-oncogenic proteins, such as mutp53 and YAPCitation42 (). In line with this notion, oncogenic RAS was recently reported to activate both SREBP1 and SREBP2, and to rely on their activity for stimulating the proliferation of transformed cells.Citation77 Likewise, the Myc oncoprotein was shown to interact with SREBP1, in a manner that enhances the transcriptional and cellular effects of Myc.Citation78 Indeed, aberrant activation of SREBP and induced expression of its target genes has been found in several cancer types, including breast, ovarian and prostate.Citation79,80 Importantly, the expression of SREBPs and their targets correlates with proliferation, invasion and cancer progression.Citation81 Accordingly, high levels of nuclear SREBP are observed in certain subtypes of glioblastoma, where they are associated with chemoresistance.Citation82 Together, these observations illustrate the importance of the SREBP network as a metabolic hub whose deregulation can drive tumorigenesis.
Statins, cancer prevention and cancer treatment
HMG-CoA reductase (HMGCR) is the rate limiting enzyme of the mevalonate pathway, and the enzymatic target of statins. In normal cells, inhibition of HMGCR with statins, and the subsequent depletion of cholesterol, triggers a robust feedback by activating SREBP. SREBP in turn transactivates 2 of its primary target genes, encoding HMGCR itself (which remains inactive due to the statins) and the LDL receptor (LDLR).Citation83 Increased levels of LDLR at the cell surface internalize circulating LDL-cholesterol, thus lowering blood cholesterol and reinstating cellular cholesterol levels. Cancer cells may become “addicted” to high cholesterol, rendering them dependent on both cholesterol import and intracellular synthesis in order to sustain sufficient levels of cholesterol. Since SREBP may be already working at maximum capacity, perhaps due to inactivation of LATS2, hyperactivation of YAP and/or mutation of p53, inhibition of HMGCR might render cancer cells particularly vulnerable to statins, despite sustained LDLR expression. Indeed, direct inhibition of SREBP1 and SREBP2 processing with fatostatin, which suppresses fatty acid and cholesterol biosynthesis, has antitumor effects in metastatic prostate cancer cells harboring mutant p53.Citation84 Furthermore, statins have been reported to possess anticancer activity in a wide range of in vitro and in vivo pre-clinical models, including liquid tumors such as leukemia and myeloma, and solid tumors such as breast, prostate, colorectal, hepatocellular, lung, pancreatic, ovarian, cervical and head and neck cancersCitation85(and references therein). For example, in a recent study involving over 1500 patients diagnosed with glioblastoma multiforme, pre-diagnostic statin treatment was associated with reduced mortality.Citation86 Similarly, in breast cancer, improved disease-free survival was found in patients taking statins before diagnosis.Citation87,88
Beyond their role in lowering cholesterol levels, statins are proposed to have other inhibitory effects on tumor growth and differentiation.Citation89,90 For example, statins may exert beneficial antineoplastic properties by altering the apoptosis-inhibitory effect of VEGF and limiting secretion of matrix metalloproteinases.Citation90,91 Unfortunately, not all attempts to determine the impact of statins on cancer have yielded unequivocal positive results.Citation92 For example, a recent study concluded that long-term (10 years) treatment with statins actually doubled the risk of invasive ductal carcinoma and invasive lobular carcinoma among postmenopausal women.Citation93 This is a disconcerting finding, given the general optimism with regard to prophylactic statin administration as a cancer prevention strategy.
Targeting the mevalonate pathway with statins might be just the tip of the iceberg. The complex regulatory network surrounding SREBP function provides a large interface for additional cancer targeting interventions. At the same time, due to feedback loops and redundant fail-safe controls, conceivably different components of the network might need to be targeted simultaneously. A better molecular understanding of SREBP regulation and deregulation, by tumor suppressors such as LATS1/2 and p53 and oncogenes such as YAP/2AZ and mutp53, might ultimately empower therapeutic manipulation of these interactions, as well as provide the foundation for better guided targeted personalized treatments with inhibitors of cholesterol biosynthesis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Prof. Uri Alon for helpful discussions and Noa Furth for insightful comments about this manuscript.
Funding
Work in the authors' lab is funded in part by a Center of Excellence grant (1779/21) from the Israel Science Foundation, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, grant 293438 (RUBICAN) from the European Research Council, the Robert Bosch Stiftung (#12.5.8000.0094.2), The German-Israeli Foundation (GIF), and the Moross Integrated Cancer Center. M.O. is incumbent of the Andre Lwoff chair in molecular biology.
References
- Silvente-Poirot S, Poirot M. Cholesterol metabolism and cancer: the good, the bad and the ugly. Curr Opin Pharmacol 2012; 12:673-6; PMID:23103112; http://dx.doi.org/10.1016/2.coph.2012.10.004
- Alikhani N, Ferguson RD, Novosyadlyy R, Gallagher EJ, Scheinman EJ, Yakar S, LeRoith D. Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene 2013; 32:961-7; PMID:22469977; http://dx.doi.org/10.1038/2nc.2012.113
- Liu J, Xu A, Lam KS, Wong NS, Chen J, Shepherd PR, Wang Y. Cholesterol-induced mammary tumorigenesis is enhanced by adiponectin deficiency: role of LDL receptor upregulation. Oncotarget 2013; 4:1804-18; PMID:24113220; http://dx.doi.org/10.18632/2ncotarget.1364
- Llaverias G, Danilo C, Mercier I, Daumer K, Capozza F, Williams TM, Sotgia F, Lisanti MP, Frank PG. Role of cholesterol in the development and progression of breast cancer. Am J Pathol 2011; 178:402-12; PMID:21224077; http://dx.doi.org/10.1016/2.ajpath.2010.11.005
- Boyd NF, McGuire V. Evidence of association between plasma high-density lipoprotein cholesterol and risk factors for breast cancer. J Natl Cancer Inst 1990; 82:460-8; PMID:2313717; http://dx.doi.org/10.1093/2nci/22.6.460
- Ferraroni M, Gerber M, Decarli A, Richardson S, Marubini E, Crastes de Paulet P, Crastes de Paulet A, Pujol H. HDL-cholesterol and breast cancer: a joint study in northern Italy and southern France. Int J Epidemiol 1993; 22:772-80; PMID:8282454; http://dx.doi.org/10.1093/2je/22.5.772
- Kitahara CM, Berrington de Gonzalez A, Freedman ND, Huxley R, Mok Y, Jee SH, Samet JM. Total cholesterol and cancer risk in a large prospective study in Korea. J Clin Oncol 2011; 29:1592-8; PMID:21422422; http://dx.doi.org/10.1200/2CO.2010.31.5200
- Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013; 342:1094-8; PMID:24288332; http://dx.doi.org/10.1126/2cience.1241908
- Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW, Mangelsdorf DJ. 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med 2007; 13:1185-92; PMID:17873880; http://dx.doi.org/10.1038/2m1641
- Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, Thompson B, Girard L, Mineo C, Brekken RA, et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep 2013; 5:637-45; PMID:24210818; http://dx.doi.org/10.1016/2.celrep.2013.10.006
- Alvarez Sanz MC, Liu JM, Huang HH, Hawrylewicz EJ. Effect of dietary protein on morphologic development of rat mammary gland. J Natl Cancer Inst 1986; 77:477-87; PMID:3461209
- de Medina P, Paillasse MR, Segala G, Voisin M, Mhamdi L, Dalenc F, Lacroix-Triki M, Filleron T, Pont F, Saati TA, et al. Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat Commun 2013; 4:1840; PMID:23673625; http://dx.doi.org/10.1038/2comms2835
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997; 89:331-40; PMID:9150132; http://dx.doi.org/10.1016/20092-8674(00)80213-5
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002; 109:1125-31; PMID:11994399; http://dx.doi.org/10.1172/2CI0215593
- Amemiya-Kudo M, Shimano H, Hasty AH, Yahagi N, Yoshikawa T, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, et al. Transcriptional activities of nuclear SREBP-1a, −1c, and −2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res 2002; 43:1220-35; PMID:12177166
- Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 1997; 99:838-45; PMID:9062340; http://dx.doi.org/10.1172/2CI119247
- Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest 1996; 98:1575-84; PMID:8833906; http://dx.doi.org/10.1172/2CI118951
- Goldstein JL, Rawson RB, Brown MS. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch Biochem Biophys 2002; 397:139-48; PMID:11795864; http://dx.doi.org/10.1006/2bbi.2001.2615
- Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev 2013; 27:355-71; PMID:23431053; http://dx.doi.org/10.1101/2ad.210773.112
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007; 21:2747-61; PMID:17974916; http://dx.doi.org/10.1101/2ad.1602907
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007; 130:1120-33; PMID:17889654; http://dx.doi.org/10.1016/2.cell.2007.07.019
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(β-TRCP). Genes Dev 2010; 24:72-85; PMID:20048001; http://dx.doi.org/10.1101/2ad.1843810
- Oren M, Aylon Y. The Hippo Signaling Pathway and Cancer. Springer Science & Business Media, New York, 2013
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014; 14:359-70; PMID:24739573; http://dx.doi.org/10.1038/2rc3711
- Aylon Y, Oren M. Chapter 1- The Paradox of p53: What, How and Why? In: G. Lozano AJL, ed. The p53 Protein: From Cell Regulation to Cancer. Cary, NC, USA: Cold Spring Harbor Laboratory Press, 2016
- Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358:15-6; PMID:1614522; http://dx.doi.org/10.1038/258015a0
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev 2006; 20:2687-700; PMID:17015431; http://dx.doi.org/10.1101/2ad.1447006
- Furth N, Bossel Ben-Moshe N, Pozniak Y, Porat Z, Geiger T, Domany E, Aylon Y, Oren M. Down-regulation of LATS kinases alters p53 to promote cell migration. Genes Dev 2015; 29:2325-30; PMID:26588988; http://dx.doi.org/10.1101/2ad.268185.115
- Kostic C, Shaw PH. Isolation and characterization of sixteen novel p53 response genes. Oncogene 2000; 19:3978-87; PMID:10962554; http://dx.doi.org/10.1038/2j.onc.1203747
- Kurinna S, Stratton SA, Coban Z, Schumacher JM, Grompe M, Duncan AW, Barton MC. p53 regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology 2013; 57:2004-13; PMID:23300120; http://dx.doi.org/10.1002/2ep.26233
- Aylon Y, Sarver A, Tovy A, Ainbinder E, Oren M. Lats2 is critical for the pluripotency and proper differentiation of stem cells. Cell Death Differ 2014; 21:624-33; PMID:24413153; http://dx.doi.org/10.1038/2dd.2013.188
- Aylon Y, Gershoni A, Rotkopf R, Biton IE, Porat Z, Koh AP, Sun X, Lee Y, Fiel MI, Hoshida Y, et al. The LATS2 tumor suppressor inhibits SREBP and suppresses hepatic cholesterol accumulation. Genes Dev 2016; 30:786-97; PMID:27013235; http://dx.doi.org/10.1101/2ad.274167.115
- Yahagi N, Shimano H, Matsuzaka T, Sekiya M, Najima Y, Okazaki S, Okazaki H, Tamura Y, Iizuka Y, Inoue N, et al. p53 involvement in the pathogenesis of fatty liver disease. J Biol Chem 2004; 279:20571-5; PMID:14985341; http://dx.doi.org/10.1074/2bc.M400884200
- Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, Ide T, Tomita S, Okazaki H, Tamura Y, et al. p53 Activation in adipocytes of obese mice. J Biol Chem 2003; 278:25395-400; PMID:12734185; http://dx.doi.org/10.1074/2bc.M302364200
- Gooley JJ, Chua EC. Diurnal regulation of lipid metabolism and applications of circadian lipidomics. J Genet Genomics 2014; 41:231-50; PMID:24894351; http://dx.doi.org/10.1016/2.jgg.2014.04.001
- Pomerening JR, Sontag ED, Ferrell JE, Jr. Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat Cell Biol 2003; 5:346-51; PMID:12629549; http://dx.doi.org/10.1038/2cb954
- Ingolia N. Cell cycle: bistability is needed for robust cycling. Curr Biol 2005; 15:R961-3; PMID:16332526; http://dx.doi.org/10.1016/2.cub.2005.11.012
- Visser S, Yang X. LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle 2010; 9:3892-903; PMID:20935475; http://dx.doi.org/10.4161/2c.9.19.13386
- Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr, Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/2PARalpha regulatory pathway. Cell Metab 2006; 4:199-210; PMID:16950137; http://dx.doi.org/10.1016/2.cmet.2006.08.005
- Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, Brown-Endres L, Tsuchihara K, Mak TW, Benchimol S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell 2011; 44:491-501; PMID:22055193; http://dx.doi.org/10.1016/2.molcel.2011.08.038
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2010; 2:a001008; PMID:20182602; http://dx.doi.org/10.1101/2shperspect.a001008
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012; 148:244-58; PMID:22265415; http://dx.doi.org/10.1016/2.cell.2011.12.017
- Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 2015; 15:73-9; PMID:25592648; http://dx.doi.org/10.1038/2rc3876
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol 2014; 16:357-66; PMID:24658687; http://dx.doi.org/10.1038/2cb2936
- Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, Zhang X, Zhang F, Chen H, Liu Y, et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci U S A 2014; 111:E89-98; PMID:24367099; http://dx.doi.org/10.1073/2nas.1319190110
- Taccioli C, Sorrentino G, Zannini A, Caroli J, Beneventano D, Anderlucci L, Lolli M, Bicciato S, Del Sal G. MDP, a database linking drug response data to genomic information, identifies dasatinib and statins as a combinatorial strategy to inhibit YAP/2AZ in cancer cells. Oncotarget 2015; 6:38854-65; PMID:26513174
- Di Agostino S, Sorrentino G, Ingallina E, Valenti F, Ferraiuolo M, Bicciato S, Piazza S, Strano S, Del Sal G, Blandino G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep 2016; 17:188-201; PMID:26691213; http://dx.doi.org/10.15252/2mbr.201540488
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009; 16:425-38; PMID:19878874; http://dx.doi.org/10.1016/2.ccr.2009.09.026
- Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, Chen J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol 2015; 17:490-9; PMID:25751139; http://dx.doi.org/10.1038/2cb3113
- Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, Lim DS, Guan KL. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol 2015; 17:500-10; PMID:25751140; http://dx.doi.org/10.1038/2cb3111
- DeRan M, Yang J, Shen CH, Peters EC, Fitamant J, Chan P, Hsieh M, Zhu S, Asara JM, Zheng B, et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep 2014; 9:495-503; PMID:25373897; http://dx.doi.org/10.1016/2.celrep.2014.09.036
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 2011; 13:376-88; PMID:21459323; http://dx.doi.org/10.1016/2.cmet.2011.03.009
- Benhamouche S, Curto M, Saotome I, Gladden AB, Liu CH, Giovannini M, McClatchey AI. Nf2/2erlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev 2010; 24:1718-30; PMID:20675406; http://dx.doi.org/10.1101/2ad.1938710
- Wan L, Sun M, Liu GJ, Wei CC, Zhang EB, Kong R, Xu TP, Huang MD, Wang ZX. Long Noncoding RNA PVT1 Promotes Non-Small Cell lung cancer cell proliferation through epigenetically regulating LATS2 expression. Mol Cancer Ther 2016; 15:1082-94; PMID:26908628; http://dx.doi.org/10.1158/2535-7163.MCT-15-0707
- Yao F, Zhou W, Zhong C, Fang W. LATS2 inhibits the activity of NF-kappa B signaling by disrupting the interaction between TAK1 and IKKbeta. Tumour Biol 2015; 36:7873-9; PMID:25946971; http://dx.doi.org/10.1007/213277-015-3362-x
- Li J, Chen X, Ding X, Cheng Y, Zhao B, Lai ZC, Al Hezaimi K, Hakem R, Guan KL, Wang CY. LATS2 suppresses oncogenic wnt signaling by disrupting β-catenin/2CL9 interaction. Cell Rep 2013; 5:1650-63; PMID:24360964; http://dx.doi.org/10.1016/2.celrep.2013.11.037
- Suzuki H, Yabuta N, Okada N, Torigata K, Aylon Y, Oren M, Nojima H. Lats2 phosphorylates p21/2DKN1A after UV irradiation and regulates apoptosis. J Cell Sci 2013; 126:4358-68; PMID:23886938; http://dx.doi.org/10.1242/2cs.125815
- Zhang K, Rodriguez-Aznar E, Yabuta N, Owen RJ, Mingot JM, Nojima H, Nieto MA, Longmore GD. Lats2 kinase potentiates Snail1 activity by promoting nuclear retention upon phosphorylation. EMBO J 2012; 31:29-43; PMID:21952048; http://dx.doi.org/10.1038/2mboj.2011.357
- Yabuta N, Mukai S, Okada N, Aylon Y, Nojima H. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle 2011; 10:2724-36; PMID:21822051; http://dx.doi.org/10.4161/2c.10.16.16873
- Tschop K, Conery AR, Litovchick L, Decaprio JA, Settleman J, Harlow E, Dyson N. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev 2011; 25:814-30; PMID:21498571; http://dx.doi.org/10.1101/2ad.2000211
- Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014; 158:833-48; PMID:25126788; http://dx.doi.org/10.1016/2.cell.2014.06.029
- Aylon Y, Yabuta N, Besserglick H, Buganim Y, Rotter V, Nojima H, Oren M. Silencing of the Lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant H-Ras-driven cell transformation. Oncogene 2009; 28:4469-79; PMID:19855428; http://dx.doi.org/10.1038/2nc.2009.270
- Aylon Y, Ofir-Rosenfeld Y, Yabuta N, Lapi E, Nojima H, Lu X, Oren M. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev 2010; 24:2420-9; PMID:21041410; http://dx.doi.org/10.1101/2ad.1954410
- Hirano Y, Yoshida M, Shimizu M, Sato R. Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J Biol Chem 2001; 276:36431-7; PMID:11477106; http://dx.doi.org/10.1074/2bc.M105200200
- Hirano Y, Murata S, Tanaka K, Shimizu M, Sato R. Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J Biol Chem 2003; 278:16809-19; PMID:12615929; http://dx.doi.org/10.1074/2bc.M212448200
- Shema E, Tirosh I, Aylon Y, Huang J, Ye C, Moskovits N, Raver-Shapira N, Minsky N, Pirngruber J, Tarcic G, et al. The histone H2B-specific ubiquitin ligase RNF20/2BRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev 2008; 22:2664-76; PMID:18832071; http://dx.doi.org/10.1101/2ad.1703008
- Kim J, Hake SB, Roeder RG. The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol Cell 2005; 20:759-70; PMID:16337599; http://dx.doi.org/10.1016/2.molcel.2005.11.012
- Shema E, Kim J, Roeder RG, Oren M. RNF20 inhibits TFIIS-facilitated transcriptional elongation to suppress pro-oncogenic gene expression. Mol Cell 2011; 42:477-88; PMID:21596312; http://dx.doi.org/10.1016/2.molcel.2011.03.011
- Lee JH, Lee GY, Jang H, Choe SS, Koo SH, Kim JB. Ring finger protein20 regulates hepatic lipid metabolism through protein kinase A-dependent sterol regulatory element binding protein1c degradation. Hepatology 2014; 60:844-57; PMID:24425205; http://dx.doi.org/10.1002/2ep.27011
- Wagner A. Energy constraints on the evolution of gene expression. Mol Biol Evol 2005; 22:1365-74; PMID:15758206; http://dx.doi.org/10.1093/2olbev/2si126
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402-12; PMID:18431400; http://dx.doi.org/10.1038/2rm2395
- Zeng L, Skinner SO, Zong C, Sippy J, Feiss M, Golding I. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell 2010; 141:682-91; PMID:20478257; http://dx.doi.org/10.1016/2.cell.2010.03.034
- Potelle S, Klein A, Foulquier F. Golgi post-translational modifications and associated diseases. J Inherit Metab Dis 2015; 38:741-51; PMID:25967285; http://dx.doi.org/10.1007/210545-015-9851-7
- Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol 2010; 17:666-72; PMID:20495563; http://dx.doi.org/10.1038/2smb.1842
- YA SNaK. Studying cell signal transduction with biomimetic point mutations. In: DF, ed. Genetic Manipulation of DNA and Protein - Examples from Current Research: InTech, 2013
- Kitano H. Cancer as a robust system: implications for anticancer therapy. Nat Rev Cancer 2004; 4:227-35; PMID:14993904; http://dx.doi.org/10.1038/2rc1300
- Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016; 35:1250-60; PMID:26028026; http://dx.doi.org/10.1038/2nc.2015.179
- Wu Y, Chen K, Liu X, Huang L, Zhao D, Li L, Gao M, Pei D, Wang C, Liu X. Srebp-1 interacts with c-Myc to enhance somatic cell reprogramming. Stem Cells 2016; 34:83-92; PMID:26388522; http://dx.doi.org/10.1002/2tem.2209
- Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care 2006; 9:358-65; PMID:16778563; http://dx.doi.org/10.1097/21.mco.0000232894.28674.30
- Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 2013; 6:1353-63; PMID:24203995; http://dx.doi.org/10.1242/2mm.011338
- Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, Nelson CC. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res 2004; 64:2212-21; PMID:15026365; http://dx.doi.org/10.1158/2008-5472.CAN-2148-2
- Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, Lin KY, Huang TT, Akhavan D, Hock MB, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2009; 2:ra82; PMID:20009104; http://dx.doi.org/10.1126/2cisignal.2000446
- Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res 2009; 50 Suppl:S15-27; PMID:18974038; http://dx.doi.org/10.1194/2lr.R800054-JLR200
- Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther 2014; 13:855-66; PMID:24493696; http://dx.doi.org/10.1158/2535-7163.MCT-13-0797
- Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene 2012; 31:4967-78; PMID:22310279; http://dx.doi.org/10.1038/2nc.2012.6
- Gaist D, Hallas J, Friis S, Hansen S, Sorensen HT. Statin use and survival following glioblastoma multiforme. Cancer Epidemiol 2014; 38:722-7; PMID:25455652; http://dx.doi.org/10.1016/2.canep.2014.09.010
- Ahern TP, Pedersen L, Tarp M, Cronin-Fenton DP, Garne JP, Silliman RA, Sorensen HT, Lash TL. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J Natl Cancer Inst 2011; 103:1461-8; PMID:21813413; http://dx.doi.org/10.1093/2nci/2jr291
- Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med 2012; 367:1792-802; PMID:23134381; http://dx.doi.org/10.1056/2EJMoa1201735
- Graaf MR, Richel DJ, van Noorden CJ, Guchelaar HJ. Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treat Rev 2004; 30:609-41; PMID:15531395; http://dx.doi.org/10.1016/2.ctrv.2004.06.010
- Denoyelle C, Vasse M, Korner M, Mishal Z, Ganne F, Vannier JP, Soria J, Soria C. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis 2001; 22:1139-48; PMID:11470741; http://dx.doi.org/10.1093/2arcin/22.8.1139
- Massaro M, Zampolli A, Scoditti E, Carluccio MA, Storelli C, Distante A, De Caterina R. Statins inhibit cyclooxygenase-2 and matrix metalloproteinase-9 in human endothelial cells: anti-angiogenic actions possibly contributing to plaque stability. Cardiovasc Res 2010; 86:311-20; PMID:19946014; http://dx.doi.org/10.1093/2vr/2vp375
- Undela K, Srikanth V, Bansal D. Statin use and risk of breast cancer: a meta-analysis of observational studies. Breast Cancer Res Treat 2012; 135:261-9; PMID:22806241; http://dx.doi.org/10.1007/210549-012-2154-x
- McDougall JA, Malone KE, Daling JR, Cushing-Haugen KL, Porter PL, Li CI. Long-term statin use and risk of ductal and lobular breast cancer among women 55 to 74 years of age. Cancer Epidemiol Biomarkers Prev 2013; 22:1529-37; PMID:23833125; http://dx.doi.org/10.1158/2055-9965.EPI-13-0414