
ABSTRACT
The TP53 protein is known to affect the sensitivity of tumor cells to cell death by DNA damaging agents. We recently reported that human and mouse cells containing an African-specific coding region variant of p53, Pro47Ser (hereafter S47), are impaired in the transactivation of a small subset of p53 target genes including GLS2 and SCO2, and are markedly resistant to cisplatin. Further, mice containing this variant are markedly predisposed to cancer. Together these findings suggested that cancer-affected humans with the S47 variant might not be effectively treated with cisplatin. To more directly test this premise, we created transformed derivatives of mouse embryo fibroblasts (MEFs) containing wild type p53 (WT) and the S47 variant and analyzed them for chemosensitivity. We find that transformation with E1A and Ras actually reverses the chemosensitivity/transcriptional differences between WT p53 and S47. Specifically, E1A/Ras-transformed S47 cells show increased sensitivity to cisplatin and paclitaxel, and comparable transactivation of GLS2 and SCO2, compared to cells with WT p53. These data suggest that the functional differences between WT p53 and S47 in primary cells may not hold true for transformed cells. They also offer hope that cisplatin and paclitaxel may be effective chemotherapeutic drugs for S47 individuals with cancer.
Introduction
The tumor suppressor gene TP53 is mutated in more than half of all human tumors. Apart from mutations that are known to alter p53 function, single nucleotide polymorphisms (SNPs) in this gene can also significantly impair p53 activity.Citation1-6 In particular, the Pro47Ser polymorphism (rs1800371 hereafter referred to as S47) has a significant influence on p53 function, by reducing phosphorylation on serine 46 and impairing p53 apoptotic and transcriptional functions.Citation7 Identifying clinical associations for this variant and cancer risk has been challenging to assess because the S47 allele is present in only 1–2% of people of African descent, and African American datasets of sufficient sample number are rare.Citation3 To overcome this challenge, we established a mouse model for this variant. We utilized the humanized p53 knock-in (Hupki) targeting allele, which replaces the mouse exons 4–9 in p53 with the corresponding human p53 exons (codon 32–332). We found that S47 mice in either homozygous or heterozygous form developed a high rate (∼75%) of spontaneous cancers, including approximately 30% with hepatocellular carcinoma.Citation8 This cancer-prone phenotype is distinct from the mouse models for Li Fraumeni syndrome, which develop sarcoma, lymphoma and some epithelial cancers.Citation9,10 Mechanistically, we found that the S47 protein was similar to WT p53 in the ability to induce apoptosis in response to most DNA damaging agents and gamma radiation, and that it was capable of transactivating the majority of p53 target genes, with the exception of 3 genes involved in cell death and metabolism: GLS2, SCO2 and PMAIP1. Additionally, we found that this variant was 10–15 fold impaired at inducing cell death by cisplatin. Interestingly, our data indicate that cisplatin induces cell death in part by inducing ferroptosis, a form of iron-mediated cell death.Citation8 Along these lines we also found that S47 cells were defective at induction of ferroptosis by erastin. These findings complement studies by others that ferroptosis is important for p53-mediated tumor suppression.Citation11 These combined findings prompted us to speculate that S47 individuals with cancer might not be effectively treated with cisplatin. In this work we sought to test this hypothesis.
Results
Generation of independent clones of WT and S47 cells transformed with E1A and Ras
To test the premise that the S47 variant might decrease the sensitivity to cisplatin and other agents in transformed cells, we infected mouse embryo fibroblasts (MEFs) containing endogenous WT p53 or the S47 variant with a retrovirus encoding E1A and Ras. To control for reproducibility we selected 2 clones of each that we confirmed have equal levels of RAS and E1A by western blot and quantitative reverse transcription-polymerase chain reaction analysis (qRT-PCR), respectively. We also compared the response of these E1A-ras transformed cells to primary WT and S47 MEFs. As expected, we saw decreased cleaved caspase 3 in primary S47 MEFs treated with cisplatin, compared to WT (). Also as expected, transformation with E1A and Ras greatly sensitized these cells to programmed cell death by cisplatin compared to primary MEFs, as assessed by protein gel blot for cleaved caspase 3. Surprisingly, however, E1A-Ras transformed WT and S47 MEFs had comparable levels of cleaved caspase 3 following cisplatin treatment, and the differences seen in primary cells were not evident in E1A-ras transformed cells ().
Figure 1. S47 MEFs transformed with E1A and Ras show increased sensitivity to cisplatin. A. Primary and E1A/Ras transformed MEFs were treated with 10 μM cisplatin for 24 hours. Cells were immunoblotted for Mdm2, p53, cleaved caspase 3, p21, Ras, and GAPDH (loading control). The data are reflective of 2 independent clones each of E1A/Ras transformed MEFs (#1 and #2). Cisplatin: CDDP. MEF: mouse embryo fibroblast. The depicted data are representative of 3 independent experiments. B. Cytotoxicity profile for cisplatin (CDDP) in WT and S47 E1A/Ras transformed MEFs. The data depicted are the average results from 2 independent clones of both WT and S47 cells, each performed in triplicate.
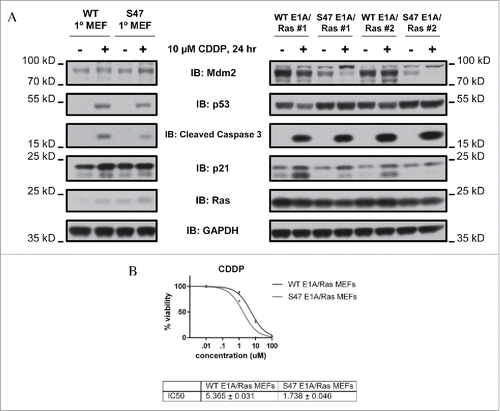
Transformation of S47 MEFs with E1A and Ras eliminates their resistance to cisplatin
We next assessed WT and S47 E1A/Ras transformed MEFs for the IC50 in for cisplatin and erastin; we showed previously that primary S47 MEFs are over 10-fold resistant to these compounds, compared to MEFs with WT p53.Citation8 In contrast to this, we found that both clones of S47 transformed cells showed at least 3-fold increased sensitivity to cisplatin, compared to WT clones (). We also found there was similar cytotoxicity to adriamycin and erastin, but increased sensitivity of S47 cells to paclitaxel (over 8-fold) (). These data suggest that transformation with E1A and Ras overrides the differences in cisplatin-mediated cytotoxicity in primary WT and S47 cells. We next analyzed transactivation of 5 p53 target genes, including 3 target genes that were differentially transactivated by p53 in WT and S47 cells: PMAIP1, GLS2 and SCO2. In primary S47 MEFs, the transactivation of GLS2 was 3-fold impaired following cisplatin treatment, compared to WT p53.Citation8 In contrast, in E1A/Ras transformed MEFs, the transactivation of GLS2 was actually greater in S47 MEFs compared to WT (5-fold versus 3-fold, respectively). Transactivation of PMAIP1 and SCO2, as well as MDM2 and p21/CDKN1A, were similar in WT and S47 E1A/Ras-infected MEFs ().
Figure 2. E1A/Ras transformed MEFs no longer show reduced transactivation of GLS2, SCO2 and NOXA (PMAIP1) in S47 cells, compared to cells with WT p53. E1A/Ras transformed MEFs were treated with 10 μM cisplatin for 24 hours. RNA was isolated and used for quantitative reverse transcription-PCR analysis, and levels were normalized to cyclophilin A.
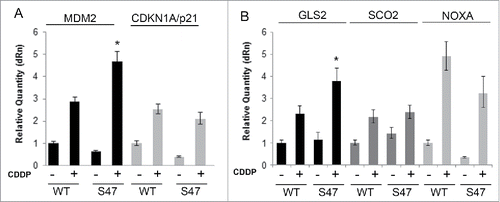
Table 1. The IC50 of cisplatin (CDDP), paclitaxel, erastin and adriamycin in WT and S47 E1A/Ras-transformed MEFs. The sulforhodamine B assay was used to determine cell viability after 72 hours of drug treatment. The data are representative of 3 independent experiments in 2 clones for each cell type, and the findings were consistent using an independent cell viability assay (trypan blue exclusion).
Concluding remarks/discussion
We previously found that both human and mouse cells containing the S47 variant of p53 were an average of 10-fold more resistant to cisplatin, and were markedly impaired for transactivation of GLS2, PMAIP1 and SCO2. We showed that the impaired transactivation of these genes accounted in part for the decreased cell death by cisplatin.Citation8 It is of note that 2 of these genes, GLS2 and SCO2, play roles in metabolism, and that the control of metabolism by p53 is implicated in tumor suppression by this protein.Citation12-14 Not surprisingly, we do find considerable differences in metabolism between cells with WT p53 and S47, with the latter showing preferred usage of aerobic glycolysis (S. Basu, unpublished data); this change in metabolism is reminiscent of the differences in metabolism between normal and tumor cells. We are currently pursuing this finding.
Based upon our data in primary mouse and human cells, we predicted that tumor cells containing the S47 protein might likewise be resistant to cisplatin, and that cisplatin treatment of S47 individuals with cancer might be poorly advised/efficacious. We therefore sought to address this important issue, as it has implications for personalized medicine treatment of S47 individuals. We were surprised to find that S47 MEFs transformed with E1A and Ras were now 3-fold more sensitive to cisplatin, and that the transcriptional differences in primary MEFs were likewise now eliminated/alleviated. The reasons for this reversal of functional differences following transformation with E1A and Ras are not entirely clear. One possibility is that the considerable stabilization and activation of p53 by E1A and Ras, even in untreated cells (), hyper-activates steady state p53 in these cells to such an extent that the transcriptional and apoptotic differences seen following cisplatin in primary cells are overall reduced. To more broadly assess the relevance of our findings, it will be of interest to determine whether other oncogene combinations lead to similar results, thus suggesting that cancer-prone individuals containing the S47 variant may in fact be favorably treated with compounds like cisplatin and paclitaxel. Indeed, we have seen decreased toxicity of cisplatin in normal kidney cells of the S47 mouse; combined with the enhanced efficacy of cisplatin in transformed cells, these data suggest that this compound may be the chemotherapeutic agent of choice in these individuals. Overall, the combined data suggest that a comprehensive analysis of the transcriptional, ferroptotic and cell death profiles of transformed WT and S47 tumor cells is warranted in order to best inform precision medicine approaches in individuals who are positive for the S47 variant.
Materials and methods
Cell culture, cytotoxicity assays, reagents
Wild-type (WT) and S47 MEFs were generated and cultured as previously described.Citation8 E1A/Ras MEFs were generated as described.Citation15 Cisplatin (CDDP; Sigma), adriamycin (Cell Signaling) erastin (Sigma), and paclitaxel (Cell Signaling) were used at the doses outlined. For cytotoxicity assays, E1A/Ras MEFs were plated at a density of 2000 cells/well in 96 well tissue culture plates and grown overnight at 37°C. Cells were treated with drugs at 10-fold increasing doses concentrations and left to incubate for 72 hrs. After 72 hrs, cell viability was assessed using the Sulforhodamine B colorimetric assay as described.Citation16
Western blotting, QRT-PCR
Total cell lysates were prepared and harvested for western blot analysis as described previously.Citation8 Antibodies used were against p53, cleaved caspase 3, and GAPDH (Cell Signaling Technology), Ras (BD Biosciences) Mdm2 (Calbiochem) and CDKN1A/p21 (sc-397, Santa Cruz Biotechnology). HRP-conjugated secondary antibodies were purchased from Jackson ImmunoResearch. For qRT-PCR analysis, RNA was isolated, converted to cDNA and used for qPCR analysis using the primers described.Citation8
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Scott Lowe (Memorial Sloan Kettering Cancer Center) for the E1A/Ras retroviral plasmid, and Donna George and Julie Leu (University of Pennsylvania) for helpful discussions.
Funding
This work was supported by R01s CA102184 and CA201430 (Murphy).
References
- Lane D. p53: out of Africa. Genes Dev 2016; 30:876-7; PMID:27083994; http://dx.doi.org/10.1101/gad.281733.116
- Azzam GA, Frank AK, Hollstein M, Murphy ME. Tissue-specific apoptotic effects of the p53 codon 72 polymorphism in a mouse model. Cell Cycle 2011; 10:1352-5; PMID:21566457; http://dx.doi.org/10.4161/cc.10.9.15344
- Basu S, Murphy ME. Genetic Modifiers of the p53 Pathway. Cold Spring Harb Perspect Med 2016; 6:469-81; PMID:27037420; http://dx.doi.org/10.1101/cshperspect.a026302
- Kang HJ, Feng Z, Sun Y, Atwal G, Murphy ME, Rebbeck TR, Rosenwaks Z, Levine AJ, Hu W. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proc Natl Acad Sci U S A 2009; 106:9761-6; PMID:19470478; http://dx.doi.org/10.1073/pnas.0904280106
- Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME. Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res 2015; 13:250-62; PMID:25256710; http://dx.doi.org/10.1158/1541-7786.MCR-14-0385
- Kung CP, Leu JI, Basu S, Khaku S, Anokye-Danso F, Liu Q, George DL, Ahima RS, Murphy ME. The P72R polymorphism of p53 predisposes to obesity and metabolic dysfunction. Cell Rep 2016; 14:2413-25; PMID:26947067; http://dx.doi.org/10.1016/j.celrep.2016.02.037
- Li X, Dumont P, Della Pietra A, Shetler C, Murphy ME. The codon 47 polymorphism in p53 is functionally significant. J Biol Chem 2005; 280:24245-51; PMID:15851479; http://dx.doi.org/10.1074/jbc.M414637200
- Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 2016; 30:918-30; PMID:27034505; http://dx.doi.org/10.1101/gad.275891.115
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119:861-72; PMID:15607981; http://dx.doi.org/10.1016/j.cell.2004.11.006
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119:847-60; PMID:15607980; http://dx.doi.org/10.1016/j.cell.2004.11.004
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015; 520:57-62; PMID:25799988; http://dx.doi.org/10.1038/nature14344
- Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 2015; 16:393-405; PMID:26122615; http://dx.doi.org/10.1038/nrm4007
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149:1269-83; PMID:22682249; http://dx.doi.org/10.1016/j.cell.2012.04.026
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011; 145:571-83; PMID:21565614; http://dx.doi.org/10.1016/j.cell.2011.03.035
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell; 88:593-602; PMID:9054499; http://dx.doi.org/10.1016/S0092-8674(00)81902-9
- Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protocols 2006; 1:1112-6; PMID:17406391; http://dx.doi.org/10.1038/nprot.2006.179