
Programmed cell death in recent years has extended to include many other cell death pathways besides apoptosis. The finding of these new cell death pathways is proving to have critical roles in how we evaluate cell death in cancer. One such cell death pathway is necroptosis, a type of programmed necrosis. Necroptosis mediates plasma membrane permeablization through phosphorylation of MLKL (mixed lineage kinase domain-like protein) by formation of the necrosome that contains RIPK3 (receptor interacting protein 3) in complex with RIPK1, FADD, and caspase-8. Death through necroptosis, but not apoptosis, promotes anti-tumor immune responses.Citation1 A role for necroptosis in cancer is further strengthened by the fact that RIPK3 expression is often silenced in cancers making them unable to undergo necroptosis.Citation2 This implies that necroptosis may be selected against during tumor evolution so that tumors can avoid adaptive anti-tumor immunity.Citation1 Moreover, apoptosis has been shown to release growth stimulating signals which could lead to more effective tumor repopulation.Citation3 Thus, apoptosis may be a less effective form of tumor cell killing than necroptosis because it is not as good at activating anti-tumor immunity, but is capable of promoting more rapid tumor repopulation. Based on these findings, it is critical to understand the underlying mechanisms that determine how a cell dies and specifically whether death occurs by apoptosis or necroptosis. A recent paper from our group provides new insights into this question.
In our recent article we show that mouse prostate cells that had lost Map3k7, a gene that is lost in 30–40% of human prostate cancers and associated with aggressive disease,Citation4 were hypersensitive to TNF and TRAIL (TNFα-related apoptosis-inducing ligand)-induced cell death.Citation5 Despite the well-established mechanism of TRAIL-induced cell death being by apoptosis, Map3k7-null cells preferentially died through necroptosis (). Moreover, the cell death mechanism could switch to apoptosis when necroptosis was prevented by direct inhibition of the necrosome. Establishing that cells lacking Map3k7 die preferentially by necroptosis in response to TRAIL could have significant immunogenic outcomes for patients whose tumors show loss of this gene. Consistent with this, and suggesting that tumors evolve ways to suppress these mechanisms, we also found that prostate cancer patients whose tumors had lost MAP3K7 also tended to have deletions of the receptors that are activated by TRAIL.
Figure 1. Cell Survival Outcomes Under TRAIL treatment: A) MAP3K7 stabilizes the ubiquitin chain of RIPK1 and allows for apoptosis. B) Loss of MAP3K7 permits RIPK1 to associate with RIPK3 which alters cell death to necroptosis. C) Additional loss of RIPK1 prevents the formation of the necrosome and therefore prevents cell death. D) Inhibition of autophagosome formation prevents necrosome activation and also prevents cell death.
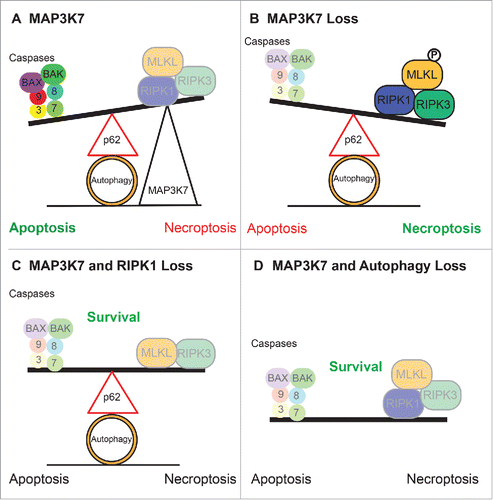
Previously, we reported TRAIL-induced apoptosis can be regulated by autophagy,Citation6 therefore we further investigated the role of autophagy in necroptosis in the Map3k7-null cells. As expected, inhibition of late stage autophagy enhanced cell death in response to TRAIL. However, inhibition of early/mid-stage autophagy by both genetic (Atg5, Atg7, and Beclin-1) and pharmacological inhibition (Wortmannin) prevented cell death. We hypothesized that components of the autophagy machinery, not the turnover of cellular components, were mediating cell death by functioning as a scaffold for necrosome complex formation and activation.
We confirmed our hypothesis by performing dual proximity ligation assays, co-immunoprecipitations, and immuno-gold transmission electron microscopy showing localization and activation of the necrosome complex on the autophagosome. Interestingly, binding of p62/SQSTM1 to RIPK1 was shown to localize the necrosome to the autophagosome, and its loss was sufficient to switch cell death from necroptosis to apoptosis. These data led us to conclude that the mechanism that determines the cell death outcome is through p62-dependent recruitment of the necrosome components to the autophagy machinery.
Our studies provide insights into how autophagy regulates necroptosis and presents a mechanism by which controlled switching between necroptosis and apoptosis can be achieved. More importantly, our studies raise a number of significant issues in regards to cancer treatment, specifically in tumors lacking Map3k7. Firstly, the use of autophagy inhibitors in the clinic could have very different outcomes based on which point of the pathway is inhibited with inhibitors that block early steps in the autophagy pathway having completely different effects than inhibitors that block later steps. Secondly, our work adds to accumulating evidence that the most important question for cancer therapy might not be just whether or not cancer cells are killed, but how they are killed. And, if adaptive immunity is activated through necroptosis, finding ways to kill cancer cells by necroptosis rather than apoptosis might be more effective. With this experimental model, where the mechanism of cell death can be switched between necroptosis and apoptosis and influenced by manipulation of autophagy, it should be feasible to test these kinds of ideas.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, Albert ML. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 2015; 350:328-34; PMID:26405229; http://dx.doi.org/10.1126/science.aad0395
- Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 2015; 25:707-25; PMID:25952668; http://dx.doi.org/10.1038/cr.2015.56
- Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O'Sullivan B, He Z, Peng Y, Tan AC, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 2011; 17:860-6; PMID:21725296; http://dx.doi.org/10.1038/nm.2385
- Rodrigues LU, Rider L, Nieto C, Romero L, Karimpour-Fard A, Loda M, Lucia MS, Wu M, Shi L, Cimic A, et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res 2015; 75:1021-34; PMID:25770290; http://dx.doi.org/10.1158/0008-5472.CAN-14-1596
- Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev Cell 2016; 37:337-49; PMID:27219062; http://dx.doi.org/10.1016/j.devcel.2016.04.018
- Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM, Thorburn A. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep 2014; 7:45-52; PMID:24685133; http://dx.doi.org/10.1016/j.celrep.2014.02.036