
Long established concepts underlying the current development of cancer therapies focus on targeting specific molecular defects that are different and unique to cancer cells; the goal being to specifically ‘shoot and kill’ the malignancy without harming the surrounding normal tissue. However in the last several years, concerns have been raised that question whether research efforts following this ideology have been truly successful in developing curative therapies versus treatments that extend life expectancy post diagnosis.Citation1
Imatinib mesylate (IM), a tyrosine kinase inhibitor (TKI), represents one of the most successful rationally-designed therapeutics for the treatment of Chronic Myeloid Leukemia (CML). Imatinib targets Bcr-Abl, the causative agent of CML and has led to dramatic clinical responses allowing CML and IM therapy to exemplify the best paradigm and justification for single target-based therapy. Despite the improvements in patient survival, we and others have demonstrated that TKIs do not kill the leukemia initiating cells (LIC) that are responsible for disease initiation and maintenance. Treated patients face the risk of developing drug resistance and most must take TKIs indefinitely with associated short and long term side effects, compliance issues and costs.Citation2 Studies implementing various kinase inhibitors, BCR-ABL knockdown and transplantation assays in a BCR-ABL mouse model suggest that CML LIC are independent of BCR-ABL kinase activity for their survival, leading us to conclude that alternative signaling pathways must maintain CML LIC.
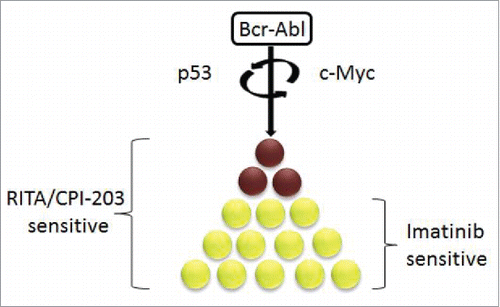
We hypothesized that a systems biology approach to identify highly connected nodes rather than single protein edges may provide deeper insight into CML biology and importantly engender a more fundamental synthetic lethality than single target approaches. By analyzing the CML proteomic signature from stem progenitor cells using network analyses, we identified that the majority of the deregulated proteins are not only connected to, but importantly dominantly regulated by, p53 and c-Myc ().Citation3
The tumor suppressor p53 is known to coordinate cellular responses to various stress factors and responds accordingly to induce apoptosis, cell cycle arrest, senescence and DNA repair. Cellular levels of p53 are tightly regulated, primarily maintained via an ubiquitin-mediated process through which the human double minute protein 2 (HDM2) is a key ubiquitin E3 ligase. As a result, much work has been undertaken to disrupt the HDM2-p53 interaction in order to stabilize p53 protein levels, and thereby rescue its often reduced function in malignant cells. Theoretically, p53 activation should lead to massive cellular death in cells dependent on oncogenes, but leave normal cells intact due to the lack of up-regulated pro-apoptotic factors within this cell population.Citation4 Due to the importance of p53 in multiple cancers, much effort has been directed to the synthesis of compounds able to stimulate its activity. One such inhibitor RITA binds p53 directly and inhibits its degradation due to its disruption of the HDM2-p53 interaction.Citation4
The MYC proto-oncogene is closely linked to growth promoting signal transduction pathways, and is one of the most highly amplified oncogenes among cancers. It is also one of the 4 important genes found to re-establish pluripotency in differentiated cells. MYC encodes a transcription factor, which can dimerize with Max and subsequently binds target DNA sequences facilitating the transcription of multiple genes involved in cell division, metabolic adaptation and survival.Citation5 The transcription of c-Myc itself is initiated by histone side chain acetylation. Histone acetylation recruits proteins with acetyl lysine binding modules or bromodomains thereby facilitating assembly of transcriptional complexes. These members of the bromodomain and extra-terminal or BET subfamily (BRD2, BRD3, BRD4, BRDT) increase the effective molarity of transcriptional activators. There are encouraging reports on the development of selective small molecule inhibitors of BET bromodomains, such as JQ1, CPI-203 and CPI-0610, which have the ability to block access of BET bromodomains to acetylated histones, through competitive inhibition, leading to cell cycle arrest and cellular senescence.Citation6,7
Our results identify that CML CD34+ cells differ from normal cells with respect to the regulation of both p53 and c-Myc above all proteins previously associated with CML.Citation3 Simultaneous targeting of these nodes in CML cells highlights a therapeutic window not previously explored. In addition our data demonstrate that TKIs do not affect the transcription of down-stream effectors. Targeting p53 and c-Myc was shown to be highly effective at eliminating the LIC that are TKI resistant, potentially explaining why TKI therapy is not curative. By obtaining a systematic overview of the CML protein signature, we are now in a position to provide a potential strategy of identifying key network modulators instrumental to the maintenance of CML cancer cell survival.
Figure 1. Bcr-Abl initiates the downregulation of the p53 tumor suppressor and the upregulation of c-Myc oncogene in CML LIC. Imatinib kills CML progenitor cells and not the tumor initiating cancer stem cells that are responsible for disease maintanance.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 2012; 12:487-93; PMID:22695393; http://dx.doi.org/10.1038/nrc3298
- Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, Legros L, Charbonnier A, Guerci A, Varet B, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010; 11:1029-35; PMID:20965785; http://dx.doi.org/10.1016/S1470-2045(10)70233-3
- Abraham SA, Hopcroft LE, Carrick E, Drotar ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT, et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016; 534:341-6; PMID:27281222; http://dx.doi.org/10.1038/nature18288
- Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 2004; 10:1321-8; PMID:15558054; http://dx.doi.org/10.1038/nm1146
- Dang CV. MYC on the path to cancer. Cell 2012; 149:22-35; PMID:22464321; http://dx.doi.org/10.1016/j.cell.2012.03.003
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011; 146:904-17; PMID:21889194; http://dx.doi.org/10.1016/j.cell.2011.08.017
- Albrecht BK, Gehling VS, Hewitt MC, Vaswani RG, Cote A, Leblanc Y, Nasveschuk CG, Bellon S, Bergeron L, Campbell R, et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J Med Chem 2016; 59:1330-9; PMID:26815195; http://dx.doi.org/10.1021/acs.jmedchem.5b01882