
Physiological functions are largely dependent on regulatory circuits. These “failsafe” molecular routes counteract the plethora of extrinsic and intrinsic stressogenic signals that threaten life forms on a daily basis. Thus, cellular and by extension organismal homeostasis is ensured. The key feature of these circuits is precise spatiotemporal organization. For instance, if a normal cell is challenged by a deleterious stimulus it will react: first, by triggering a “red alert” mobilizing an array of responses that ultimately aim in neutralizing the toxic factor(s) and second, when “all is clear” by returning in the “status quo ante” (previous normal) condition. The later is accomplished through negative feedback signals that “switch OFF” the alarm.Citation1 Thus, when the p53 network is intact, and the normal cell is challenged, p21WAF1/Cip1 activation occurs temporary leading to a short-term cell cycle arrest providing time for the cellular repair mechanisms to deal with the threat. Once the danger is over, MDM2 another direct p53 target “switches OFF” p53 by degrading it (). Within the same line, p21WAF1/Cip1 is only transiently expressed during the acute phase of senescence until the p16INK4/pRB pathway takes over to establish the irreversible senescent state (chronic phase).Citation2 In pathophysiological situations, like cancer, this accurate spatiotemporal organization is largely disrupted and new signaling routes are set up defying in most cases the “failsafe” process. A characteristic paradigm of this deregulated process is the constitutive activation of the DNA damage response checkpoint(s) in cancer.Citation3 Likewise, in a p53-deficient cancerous milieu the p53-p21WAF1/Cip1-MDM2 failsafe circuit is non-functional leaving CDKN1A (gene encoding p21WAF1/Cip1) responsive to a wide spectrum of p53-independent stimuli, most of which are well established cancer promoting factors such as, FGF2, EGF and IL-6.Citation4
Figure 1. Physiological and pathological actions of p21WAF1/Cip1. In a physiological regulatory circuit p21WAF1/Cip1 is the prototype downstream-effector of wild-type (wt) p53 exerting short-term effect essential for replication and error-free repair. Chronic p21WAF1/Cip1 induction in a p53-null environment fuels genomic instability. See text for details. Abbreviations: TLS: translesion DNA synthesis, DSB: DNA: double strand breaks.
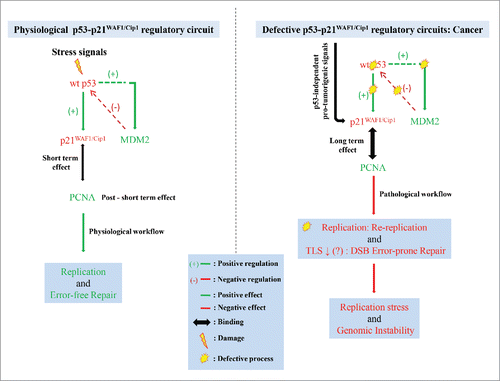
Within this context we demonstrated that the duration of p21WAF1/Cip1 expression becomes longer (chronic), as opposed to the short-term p53-mediated induction (transient cell cycle arrest), leading to an initial senescent phase that a first glance appears as an efficient anti-proliferative and eventually anti-tumor response.Citation5 Although cellular senescence is considered an anti-tumor barrier, in reality it is an “imperfect” tumor-suppressive mechanism. The prevailing view for the later notion is the so called senescence-associated secretory phenotype (SASP), a process that exercises pro-tumorigenic effects.Citation2 We found out, however, that p21WAF1/Cip1-driven senescence conceals a more complex and “malicious” program characterized by S-M phase dissociation, due to mitotic blockage and over-replication in the form of re-replication, as a result of deregulated licensing.Citation5 The key biochemical event leading to aberrant licensing is the interaction of p21WAF1/Cip1 with Proliferating Cell Nuclear Antigen (PCNA), a DNA sliding clamp involved in fundamental cell cycle functions such as, DNA replication and repair. Among all PCNA-interacting partners, p21WAF1/Cip1 possesses the highest binding affinity (KD ∼2.5 nM), implying that under conditions of “chronic” expression, p21WAF1/Cip1 can displace all other PCNA competitors. In line with this notion, we showed that protracted p21WAF1/Cip1 expression abrogated the property of PCNA to regulate the degradation cycle of the replication licensing factors Cdt1 and Cdc6,Citation6 triggering re-replication.Citation5 Consecutively, p21WAF1/Cip1-driven re-replication causes replication stress that acts as a selective process leading eventually to senescence over-ride and the “birth” of more aggressive off-springs termed “escaped cells,” conferring to tumor heterogeneity. The selective process is actually an extensive error-prone repair process that ensures that over time the “fittest and more stable cells” will survive and emerge.Citation5 An essential biochemical act assisting in p21WAF1/Cip1 senescence bypass is suppression of the INK4/ARF locus by the deregulated replication licensing factor Cdc6,Citation6 rendering the senescent phase reversible.Citation2
Another consequence of continuous p21WAF1/Cip1 expression, related to PCNA-dependent functions, could be suppression of PCNA mono-ubiquitinilation (data not shown), essential for translesion DNA synthesis (TLS) and repair.Citation4 Since TLS is responsible for the majority of single-nucleotide-substitutions (SNSs) observed in cancer,Citation7 a prediction is that the p21WAF1/Cip1-mediated “escaped” cells should exhibit a lower SNS “load” relative to non-induced cells. Indeed analyzing the data in our report, we found out that this was the case.Citation5 TLS is the main DNA damage tolerance pathway of the cell representing an important “defense” line, as it mends the lesions that escape from the surveillance of the error-free DNA repair mechanisms, allowing completion of chromosome replication.Citation7 Apparently, prolonged p21WAF1/Cip1 expression could lead to reduced TLS activity contributing together with re-replication to replication fork stalling, collapse and generation of double strand breaks fuelling genomic instability (). However, the biological significance of the later scenario remains to be proven.
At the end of the day losing wild-type p53 activity is like opening the Pandora's box with p21WAF1/Cip1 being a “new evil” released.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Rué P, Garcia-Ojalvo J. Modeling gene expression in time and space. Annu Rev Biophys 2013; 42:605-27; PMID:23527779; http://dx.doi.org/10.1146/annurev-biophys-083012-130335
- Campisi J, d'Adda diFagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8(9):729-40; PMID:17667954; http://dx.doi.org/10.1038/nrm2233
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319(5868):1352-5; PMID:18323444; http://dx.doi.org/10.1126/science.1140735
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009; 9(6):400-14; PMID:19440234; http://dx.doi.org/10.1038/nrc2657
- Galanos P, Vougas K, Walter D, Polyzos A, Maya-Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gags S, Tzetis M, et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 2016; 18(7):777-89; PMID:27323328; http://10.1038/ncb3378
- Petrakis TG, Komseli E-S, Papaioannou M, Vougas K, Polyzos A, Myrianthopoulos V, Mikros E, Trougakos IP, Thanos D, Branzei D, et al. Exploring and exploiting the systemic effects of deregulated replication licensing. Semin Cancer Biol 2016; 37–38:3-15; PMID:26707000; http://dx.doi.org/10.1016/j.semcancer.2015.12.002
- Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res 2013; 2(3):107-129; PMID: 24058901; http://dx.doi.org/10.3978/j.issn.2218-676X.2013.04.01