
ABSTRACT
Alterations in cell cycle regulation underlie the unrestricted growth of neoplastic astrocytes. Chemotherapeutic interventions of gliomas have poor prognostic outcomes due to drug resistance and drug toxicity. Here, we examined the in vitro growth kinetics of C6 glioma (C6G) cells and primary astrocytes and their responses to 2 phase-specific inhibitors, lovastatin and hydroxyurea. C6G cells demonstrated a shorter G1 phase and an earlier peak of DNA synthesis in S phase than primary astrocytes. As C6G cells and primary astrocytes re-entered the cell cycle in the presence of lovastatin or hydroxyurea, they exhibited different sensitivities to the inhibitory effects of these agents, as measured by [3H]-thymidine incorporation. Compared to primary astrocytes, C6G cells were more sensitive to lovastatin, but less sensitive to hydroxyurea. Studies using 2 different paradigms of exposure uncovered dramatic differences in the kinetics of DNA synthesis inhibition by these 2 agents in C6G cells and primary astrocytes. One notable difference was the ability of C6G cells to more easily recover from the inhibitory effects of hydroxyurea following short exposure. Our results provide insight into C6 glioma drug resistance as well as the inhibitory effects of these 2 phase-specific inhibitors and their chemotherapeutic potential.
Introduction
Under normal circumstances, proliferation of mammalian astrocytes accompanies prenatal and early postnatal brain development. Astrocytes then enter a state of quiescence (G0 phase); re-entry into the cell cycle can be stimulated by reactive processes or neoplastic transformation.Citation1-6 The mechanisms involved in malignant transformation and subsequent neoplastic proliferation of mammalian astrocytes that lead to the formation of gliomas are poorly understood. However, they are believed to be related to altered signal transduction, loss of apoptosis, or cell cycle dysregulation.Citation7-10 As a result of our lack of understanding of the molecular mechanisms of gliomas, rates of recurrence and mortality are high in patients with different types of gliomas, despite therapeutic interventions.Citation11-13
Our understanding of the regulatory mechanisms underlying the transition from G1 to S phase in eukaryotic cells is incomplete. Studies have implicated different protein families in G1 restriction point regulation, such as the cdc25 phosphatase family, the cyclin family, and the retinoblastoma (Rb) protein.Citation14,15 In addition, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, which converts 3-HMG-CoA into mevalonate and downstream isoprenoids in the mevalonate pathway, has been shown to be essential in regulating G1/S phase progression in mammalian cells, including astrocytes.Citation16-18 Depletion of mevalonate with lovastatin, a competitive inhibitor of HMG-CoA reductase, was shown to result in G1 phase cell cycle arrest.Citation19 Depletion of mevalonate and isoprenoids via statin-mediated inhibition of HMG-CoA reductase has been shown to decrease the expression or function of cell cycle proteins, and has been suggested as a potential strategy for the prevention or treatment of cancers.Citation20
Biochemical abnormalities and alterations in HMG-CoA reductase activity have been documented in C6 gliomas (C6G) and in brain tumors, respectively.Citation21,22 In addition, it was reported that the mevalonate pathway was essential for the proliferation of human and rodent glioma cells, as shown through inhibition of the HMG-CoA reductase by simvastatin.Citation23,24 Accordingly, HMG-CoA reductase has long been considered a potential target for the treatment of gliomas. We previously demonstrated that, following cell cycle arrest and subsequent re-entry, C6G cells exhibit distinctive in vitro growth kinetics as compared to newborn and adult rat astrocytes.Citation25 However, the differential effects of phase-specific cell cycle inhibitors on the growth kinetics of C6G cells versus their untransformed counterparts (i.e. primary astrocytes) have not yet been examined.
In the present study, we examined the in vitro effects of 2 common phase-specific inhibitors, lovastatin (i.e., G1 specific) and hydroxyurea (i.e. S specific), on the proliferation of C6G cells and primary rat astrocytes following serum deprivation and subsequent serum up-shift. We also examined the differential effects of the 2 phase-specific inhibitors on these cells as they progressed through the cell cycle, utilizing brief exposure paradigms of both delayed addition and early removal of the inhibitors when the in vitro cultures re-entered the cell cycle.
Methods
Animals and materials
Adult Sprague-Dawley rats were provided by the Animal Research Center, SUNY at Buffalo. Newborn pups were purchased from Harlan Sprague Dawley. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the SUNY at Buffalo. The C6G cell line was purchased from ATCC (Product # ATCC® CCL-107™; http://www.atcc.org/products/all/CCL-107.aspx). Dulbecco's Modified Eagles medium (DMEM) was purchased from Gibco/Life Technologies (Product # 11965-092; https://www.thermofisher.com/order/catalog/product/11965092). Fetal bovine serum (FBS; Product # SH30071.03HI; https://promo.gelifesciences.com/gl/hyclone/product/hyclone-fetal-bovine-serum-u-s-characterized.html) and bovine calf serum (BCS; Product # SH30073.03HI; https://promo.gelifesciences.com/gl/hyclone/product/hyclone-calf-serum-u-s.html) were purchased from Hyclone. Tissue culture flasks and other disposable products for cell culture were obtained from either Corning (Product #3276; http://catalog2.corning.com/LifeSciences/en-US/Shopping/ProductDetails.aspx?categoryname=andproductid=3276(Lifesciences)) or Becton Dickinson (Product # 08-772-33; https://www.fishersci.com/shop/products/falcon-tissue-culture-plates-23/p-154828). Nylon mesh was purchased from Small Parts and fashioned into pouches for cell dissociation. Tritiated [methyl3H] thymidine (6.7 Ci/mmol) was purchased from Amersham, while Bradford protein reagent was purchased from BioRad (Product # 5000002; http://www.bio-rad.com/en-us/product/bio-rad-protein-assay). Scintillation fluid (EcosintA) was obtained from National Diagnostics (Product # LS-273; https://www.nationaldiagnostics.com/liquid-scintillation/product/ecoscint). Lovastatin was a generous gift of Merck, Sharpe, and Dohme. Hydroxyurea and other reagents were purchased from Sigma-Aldrich.
Rat astrocyte culture
Primary rat astrocyte cultures were generated by mechanical dissociation of cerebral cortices of newborn Sprague-Dawley rats aged <36 hours as previously described.Citation17 Cultures were maintained in DMEM and 10% FBS in 5% CO2/95% humidified air at 37°C.Citation1,3 At 10–14 days, primary cultures were passaged at a density of 104cells/cm2. The resulting cultures were given fresh medium every 5 d until confluence. Final astrocyte cultures were >95 % pure based on their immunoreactivity with anti-glial fibrillary acid protein antibody.Citation1
C6G culture
The C6G cultures were grown in DMEM and 10% FBS according to previously described procedures.Citation25,26 Cultures were maintained in 5% CO2/95% humidified air at 37°C. Initial cultures were passaged at a concentration of 2,000 cells/cm2. All experiments were performed on confluent cultures.
In vitro serum deprivation and serum up-shift
All experiments with the C6G cells and primary astrocyte cultures were performed in parallel using the same batches of media and agents. After reaching confluence, both C6G cell and primary astrocyte cultures were subcultured into 6-well plates at their aforementioned concentrations, using a protocol of sequential enzymatic and mechanical disruption as previously described.Citation17,25,26 Both cell types were allowed to grow to 30% to 50% confluence (5-7 d and 1-3 d after passage of primary astrocytes and C6G cells, respectively, in 10% BCS/DMEM). At that point, the culture medium was removed, the cells were washed with warm PBS (pH 7.4), and the cells were overlaid with DMEM plus 0.1% BCS. The cells were left in the serum-depleted medium for 48 hours, forcing the cells into cell cycle arrest. After 48 hours of serum deprivation, the cells were re-exposed to DMEM plus 10% BCS, allowing re-entry of the cells into the cell cycle. The time of serum up-shift is considered to be the start of the experiment. Based on BrdU pulse labeling experiments, this protocol renders approximately 85% of the cells into cell cycle arrest.Citation26 To this date, flow cytometric analysis has been unsuccessful in primary astrocytes due to the heterogeneous nature of primary astrocyte size and shape,Citation26 even though it has been used to confer the G0 phase of the cell cycle phase in certain mammalian cells. Therefore, the use of FACS analysis to study primary astrocytes remains a major technical challenge.
Assay of cell proliferation
The incorporation of tritiated [methyl-3H]-thymidine into C6G cells and primary astrocytes was used to quantify cell proliferation.Citation17,26 One hour prior to termination of the incubation period, [3H]-thymidine was added to the wells (final activity: 1 µCi/ml of medium), and the cells were returned to 5% CO2/95% humidified air at 37°C for the final hour of incubation. Supernatants in all wells were then aspirated and cells were washed with PBS (pH 7.4) twice to remove excess [3H]-thymidine. DNA and total cellular proteins were precipitated with trichloroacetic acid.Citation3,17 Cell proliferation was measured as incorporation of radioactivity per microgram of protein present in the acid-precipitated portion (cpm/μg protein). The samples were counted for tritium in a β counter (LKB Wallac) for 10 minutes. Protein concentrations were determined by the Bradford Assay using a BioRad Microplate reader (Model 3550-UV) at wavelength of 595 nm.
Phase-specific inhibition of the cell cycle
Following the in vitro serum deprivation and subsequent serum up-shift protocol, C6G cells and primary astrocytes were treated with either the G1-phase specific inhibitor, lovastatin, or the S-phase specific inhibitor, hydroxyurea. In the first set of experiments, these cells were incubated with graded concentrations of either one of these 2 specific inhibitors as they entered the cell cycles. The rates of proliferation in these cells following incubation with the phase-specific inhibitors were determined at the peak of their respective S phases. In addition to the proliferation assays, parallel studies were also performed in 4-chambered slides to determine the cell survival by using trypan blue staining. In the second set of experiments, these cells were incubated with lovastatin at a final concentration of 10−5 M, or hydroxyurea at a final concentration of 10−3 M for variable amounts of time as defined below. ‘Delayed addition’ and ‘early removal’ protocols () were utilized to study the kinetics of cell cycle progression in both cell types.
Figure 1. Experimental paradigm for “early removal” and “delayed addition” of lovastatin or hydroxyurea to the culture media of C6G cells or primary astrocytes.
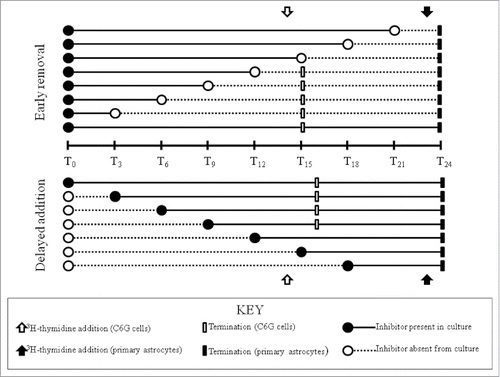
Delayed addition paradigm
Inhibitors were added to the cultures at different time points after serum up-shift (i.e., at 3 hours, 6 hours, etc in 3 hour intervals). Peak DNA synthesis occurs in C6G cells at 15 hours and in primary astrocytes at 24 hours; based on this finding, [3H]-thymidine was added to both cell cultures one hour prior to peak DNA synthesis (final activity: 1 µCi/ml). Incubation of both cell cultures was terminated one hour later and the cells were processed to determine rate of cell proliferation as described above.
Early removal paradigm
Inhibitors were added to the cultures at serum up-shift, and the medium containing the inhibitor was removed at different time points after this (i.e. at 3 hours, 6 hours, etc in 3 hour intervals). The cell cultures were then washed with PBS twice to ensure complete removal of the inhibitor. Cell cultures were overlaid with fresh medium that was free of inhibitor and incubated until the peak of DNA synthesis, i.e., at 15 hours for C6G cells and at 24 hours for primary astrocytes. One hour prior to termination, [3H]-thymidine was added to both cell cultures (final activity: 1 µCi/ml). The cells were then processed to determine rate of cell proliferation as described above.
Statistical analysis
All data are presented as a mean ± standard error of the mean (SEM). Statistical analysis was performed using the student's t-test for paired samples. A p value of less than 0.05 was considered to be statistically significant.
Results
Cell cycle kinetics following serum up-shift
We previously found that C6G cells and primary astrocytes exhibit different in vitro growth kinetics in response to serum depletion and serum up-shift.Citation25 We set out to confirm these differences with current batches of cell stocks and lots of culture media, sera, and reagents to minimize any confounding variables that might influence the results of adding phase-specific agents to the cultures. The emergence of the C6G cells from G1 to undergo active DNA synthesis occurred 6 hours after serum up-shift; in contrast, primary astrocytes did not enter S phase until approximately 12 hours after serum up-shift (). Despite the earlier S-phase entry by C6G cells, the slope of the rise in DNA synthesis remained similar in both cell types. Finally, the peak of DNA synthesis occurred earlier in C6G cells (15 hours) than in primary astrocytes (22 hours).
Figure 2. Kinetics of DNA synthesis in C6G cells and primary astrocytes following serum deprivation and serum up-shift. C6G cells and primary astrocytes were first serum-deprived for 48 hours, then allowed to re-enter the cell cycle through serum up-shift. [3H]-thymidine was added to both cell cultures at different time points after serum up-shift for one hour prior to termination. Cell proliferation in both cell cultures was measured by the incorporation of [3H]-thymidine into the DNA of both cell cultures. All data points represent the mean ± SEM of at least 2 experiments.
![Figure 2. Kinetics of DNA synthesis in C6G cells and primary astrocytes following serum deprivation and serum up-shift. C6G cells and primary astrocytes were first serum-deprived for 48 hours, then allowed to re-enter the cell cycle through serum up-shift. [3H]-thymidine was added to both cell cultures at different time points after serum up-shift for one hour prior to termination. Cell proliferation in both cell cultures was measured by the incorporation of [3H]-thymidine into the DNA of both cell cultures. All data points represent the mean ± SEM of at least 2 experiments.](/cms/asset/19300469-cc39-4e04-8e6f-3c367542079b/kccy_a_1220454_f0002_b.gif)
Effects of phase specific inhibitors on cell proliferation
To investigate the effects of 2 different phase-specific inhibitors on DNA synthesis following serum up-shift, C6G cells and primary astrocyte cultures were allowed to proliferate in the presence of graded concentrations of a phase-specific inhibitor for 15 hours and 24 hours, respectively. Treatment with both lovastatin, a G0/G1 phase-specific inhibitor, and hydroxyurea, an S phase inhibitor that blocks DNA synthesis by interacting with nucleotide reductase, resulted in inhibition of DNA synthesis in C6G cells and primary astrocytes in concentration-dependent manners (). C6G cells displayed greater sensitivities to the inhibitory effects of lovastatin compared to primary astrocytes, as shown by an upward shift in the concentration-effect curve (). These data support the notion that mevalonate availability is critical for DNA replication in both cell types during cell cycle progression, particularly C6G cells. The inhibitory effects of hydroxyurea were significantly greater in primary astrocytes at all concentrations tested (p < 0.001), as demonstrated by a downward shift of the concentration-effect curve of C6G cells vs. that of primary astrocytes (). At the highest concentration of hydroxyurea tested (1 mM), the percent inhibition of DNA synthesis was 64.08 ± 2.10% in C6G cells and 81.02 ± 3.65% in primary astrocytes. This contrasts the more pronounced inhibitory effects of lovastatin on C6G cells than primary astrocytes (66.84 ± 2.40% vs. 54.33 ± 2.66%, respectively; 10 μM lovastatin). These inhibitory effects were not results of cell death caused by the phase-specific inhibitors, as parallel studies using trypan blue staining revealed minimal amount of cell death at the concentrations of the phase-specific inhibitors tested (overall 1-3% in both groups) (data not shown).
Figure 3. Effects of lovastatin (a) and hydroxyurea (b) on DNA synthesis in C6G cells and primary astrocytes. C6G cells and primary astrocytes were first serum-deprived for 48 hours, then allowed to re-enter the cell cycle through serum up-shift in the presence of graded concentrations of lovastatin and hydroxyurea. [3H]-thymidine was added to the medium one hour prior to the peak of DNA synthesis in both cell cultures (15 hours for C6G cells, 24 hours for primary astrocytes). Incubation of both cell cultures was terminated one hour later, and cell proliferation was measured by the incorporation of [3H]-thymidine into the DNA. All data points are presented as mean ± SEM of three experiments. : p < 0.05 for 10−9 M; p < 0.001 for 10−7 M; and p < 0.01 for 10−6 M and 10−5 M. 10i9 M, not significant. : p < 0.001 for all data points.
![Figure 3. Effects of lovastatin (a) and hydroxyurea (b) on DNA synthesis in C6G cells and primary astrocytes. C6G cells and primary astrocytes were first serum-deprived for 48 hours, then allowed to re-enter the cell cycle through serum up-shift in the presence of graded concentrations of lovastatin and hydroxyurea. [3H]-thymidine was added to the medium one hour prior to the peak of DNA synthesis in both cell cultures (15 hours for C6G cells, 24 hours for primary astrocytes). Incubation of both cell cultures was terminated one hour later, and cell proliferation was measured by the incorporation of [3H]-thymidine into the DNA. All data points are presented as mean ± SEM of three experiments. Fig. 2a: p < 0.05 for 10−9 M; p < 0.001 for 10−7 M; and p < 0.01 for 10−6 M and 10−5 M. 10i9 M, not significant. Fig. 2b: p < 0.001 for all data points.](/cms/asset/8f459e31-5892-403d-add8-1b78ab739da9/kccy_a_1220454_f0003_b.gif)
Brief exposure to lovastatin: Delayed addition and early removal
In order to examine the effects of the 2 phase-specific inhibitors on proliferating astrocytes or gliomas, the cell cultures were studied using a paradigm of brief exposure to the 2 phase-specific inhibitors. The effect of delayed lovastatin exposure on DNA synthesis was examined by adding lovastatin to cultures at varying intervals after serum up-shift. All data were expressed as percent of the maximal inhibition of DNA synthesis in each group (i.e., inhibition at a lovastatin concentration of 1 × 10−5 M, or a hydroxyurea concentration of 1 × 10−3 M, added at serum up-shift; see ) or percent of maximum DNA synthesis in each group. In primary astrocytes, the delayed addition of lovastatin after serum up-shift led to a more gradual reduction in DNA synthesis inhibition, such that 50% inhibition (IC50) did not occur until around 8 hours (). In C6G cells, maximal inhibition of DNA synthesis was significantly blocked when lovastatin was added 3 hours after the serum up-shift. More than 80% of DNA synthesis inhibition in C6G cells by lovastatin occurred within the first 3 hours of treatment. Therefore, there was a dramatic shift of the IC50 to a much earlier time point in C6G cells (∼2 hours), as compared to primary astrocytes (∼8 hours). Since HMG-CoA reductase is believed to play an important role in G1 to S phase cell cycle progression, the early sensitivity to the inhibitory effects of lovastatin observed in the C6G cells might be inherent to shorter baseline kinetics leading up to DNA synthesis in these cells ().
Figure 4. Effect of a) delayed addition to and b) early removal of lovastatin from cell cultures on DNA synthesis of primary astrocytes and C6G cells. Serum deprived C6G cells and primary astrocytes were allowed to re-enter the cell cycle through serum up-shift. a) At various time points after serum up-shift, lovastatin was added to both cultures (final concentration: 10−5 M). [3H]-thymidine was added to culture medium one hour prior to the peak of DNA synthesis in both cell cultures (15 hours for C6G cells, 24 hours for primary astrocytes). Incubation of both cell cultures was terminated one hour later. b) At different time points after serum up-shift, medium containing lovastatin was removed. Cell cultures were then overlaid with medium that was free of lovastatin and incubated until the peak of DNA synthesis (15 hours for C6G cells, 24 hours for primary astrocytes). One hour prior to termination, [3H]-thymidine was added to both cell cultures. For both a) and b), effects of lovastatin on DNA synthesis of both cultures at different time points were compared to untreated controls (i.e., maximum DNA synthesis). All data points represent results from 2 experiments.
![Figure 4. Effect of a) delayed addition to and b) early removal of lovastatin from cell cultures on DNA synthesis of primary astrocytes and C6G cells. Serum deprived C6G cells and primary astrocytes were allowed to re-enter the cell cycle through serum up-shift. a) At various time points after serum up-shift, lovastatin was added to both cultures (final concentration: 10−5 M). [3H]-thymidine was added to culture medium one hour prior to the peak of DNA synthesis in both cell cultures (15 hours for C6G cells, 24 hours for primary astrocytes). Incubation of both cell cultures was terminated one hour later. b) At different time points after serum up-shift, medium containing lovastatin was removed. Cell cultures were then overlaid with medium that was free of lovastatin and incubated until the peak of DNA synthesis (15 hours for C6G cells, 24 hours for primary astrocytes). One hour prior to termination, [3H]-thymidine was added to both cell cultures. For both a) and b), effects of lovastatin on DNA synthesis of both cultures at different time points were compared to untreated controls (i.e., maximum DNA synthesis). All data points represent results from 2 experiments.](/cms/asset/32b72389-6e1d-43f9-8eb3-b0b98c338d6d/kccy_a_1220454_f0004_b.gif)
To evaluate the effects of lovastatin on the magnitude of DNA synthesis in both cell types, cultures were briefly exposed to lovastatin at intervals from the time of serum up-shift to a maximum of 12 hours in C6G cells or 21 hours in primary astrocytes. All data were expressed as a percent of the maximum DNA synthesis seen in controls not exposed to lovastatin. A 3-hour exposure to lovastatin at serum up-shift led to an appreciable reduction in DNA synthesis in primary astrocytes, resulting in only 51% of maximum DNA synthesis (). Extending the exposure time to 21 hours resulted in an additional 10% decrease in DNA synthesis. C6G cells also exhibited a dramatic reduction (60% of maximum DNA synthesis) after a 3 hour exposure. However, in contrast to primary astrocytes, an extended exposure (up to 12 hours) led to a more substantial reduction in DNA synthesis, resulting in only 29% of maximum DNA synthesis. Therefore, C6G cells were more likely to recover from the inhibitory effects of lovastatin () following short exposure (i.e. 3 hours) as opposed to longer exposure (i.e., 12 hours), while lovastatin exposure only moderately impacted the ability of primary astrocytes to recover following longer exposure (i.e. 12 hours) versus short exposure (i.e., 3 hours).
Brief exposure to hydroxyurea: Delayed addition and early removal
By using a similar paradigm of brief exposure to a phase-specific inhibitor, the inhibitory effects of hydroxyurea on both cell cultures were examined. Delayed addition of hydroxyurea was performed according to a protocol identical to that used for lovastatin. The results revealed that inhibition of DNA synthesis was significantly increased in primary astrocytes when hydroxyurea was added at 9 hours and 12 hours (119.70 ± 2.73% and 121.43 ± 1.51%, respectively, vs. 100% at serum up-shift). When hydroxyurea was added after 12 hours, inhibition of DNA synthesis was significantly reduced as compared to when it was added at serum up-shift (). C6G cells also demonstrated increased inhibition of DNA synthesis when the addition of hydroxyurea occurred at 6 hours (117.10 ± 2.84%). Hence, inhibition of DNA synthesis initially increases above the control levels in both primary astrocytes (6 hours – 12 hours) and C6G cells (6 hours), and declines thereafter. Furthermore, hydroxyurea inhibited DNA synthesis more effectively when added immediately before the S phase. Overall, C6G cells demonstrated a similar kinetic response pattern to that of primary astrocytes, but responded more rapidly.
Figure 5. Effect of a) delayed addition to and b) early removal of hydroxyurea from to cell cultures on DNA synthesis of C6G cells and primary astrocytes. Serum deprived C6G cells and primary astrocytes were allowed to re-enter the cell cycle through serum up-shift. a) At various time points after serum shift-up, hydroxyurea was added to both cultures (final concentration: 10−3 M). [3H]-thymidine was added to both cell cultures one hour prior to the peak of DNA synthesis (15 hours for C6G cells, 24 hours for primary astrocytes). Incubation of both cell cultures was terminated one hour later. b) At different time points after serum up-shift, medium containing hydroxyurea was removed. Cell cultures were then overlaid with medium that was free of hydroxyurea and incubated until the peak of DNA synthesis (24 hours for primary astrocytes, 15 hours for C6G cells). [3H]-thymidine was added to both cell cultures one hour prior to termination. For both a) and b), effects of hydroxyurea on DNA synthesis of both cultures at different time points were compared to that at T0 (i.e. maximum inhibition). All data points represent results from 2 experiments.
![Figure 5. Effect of a) delayed addition to and b) early removal of hydroxyurea from to cell cultures on DNA synthesis of C6G cells and primary astrocytes. Serum deprived C6G cells and primary astrocytes were allowed to re-enter the cell cycle through serum up-shift. a) At various time points after serum shift-up, hydroxyurea was added to both cultures (final concentration: 10−3 M). [3H]-thymidine was added to both cell cultures one hour prior to the peak of DNA synthesis (15 hours for C6G cells, 24 hours for primary astrocytes). Incubation of both cell cultures was terminated one hour later. b) At different time points after serum up-shift, medium containing hydroxyurea was removed. Cell cultures were then overlaid with medium that was free of hydroxyurea and incubated until the peak of DNA synthesis (24 hours for primary astrocytes, 15 hours for C6G cells). [3H]-thymidine was added to both cell cultures one hour prior to termination. For both a) and b), effects of hydroxyurea on DNA synthesis of both cultures at different time points were compared to that at T0 (i.e. maximum inhibition). All data points represent results from 2 experiments.](/cms/asset/25a7256a-1340-49e4-a606-f2f82c4c5676/kccy_a_1220454_f0005_b.gif)
The early removal paradigm for hydroxyurea produced quite different results (). DNA synthesis in primary astrocytes only reached 55.21% of its zenith when hydroxyurea was removed 3 hours after serum up-shift; a similar value was seen with 6 hours of exposure to this inhibitor (57.31 ± 1.96%). After that point, the reduction in DNA synthesis continued with a nadir of 38.04 ± 0.41% of control when hydroxyurea was removed at 21 hours. Therefore, shortening the exposure time of primary astrocytes from 21 to 3 hours led to a loss of inhibition of DNA synthesis of 17%, which was about 20% of the maximum inhibition seen at 24 hours. In contrast, C6G cells exhibited only a slight decrease in DNA synthesis after a 3-hour exposure (94.04% of maximum DNA synthesis in the control). Prolonging the exposure time led to an additional reduction in DNA synthesis (55.42 ± 0.51% of maximum DNA synthesis following an exposure of 12 hours). Of note, shortening the exposure of C6G cells to hydroxyurea from 15 hours (i.e. maximum inhibition) to 3 hours led to a loss of inhibition of approximately 58%, which was about 90% of the maximum inhibition, and 4.5 times higher than that seen in primary astrocytes. Thus, the delayed addition and early removal protocols reveal that, in contrast to the enhanced sensitivities of the C6G cells to lovastatin (), C6G cells were more resistant to brief hydroxyurea exposure than primary astrocytes and were also better able to recover from DNA synthesis inhibition induced by these conditions.
Discussion
Despite years of research, prognostic outcomes for gliomas remain poor due to drug resistance, drug toxicity, and an incomplete understanding of the biochemical and cellular alterations that lead to the neoplastic transformation of glia. Among the essential differences between neoplastic and non-neoplastic cells is the capacity of the former to divide indefinitely; the mechanisms underlying this uncontrolled growth are under intensive study. However, examining cell cycle regulation of both neoplastic and non-neoplastic glia in vivo is difficult due to a plethora of technical challenges; for example, normal and tumoral glia may be asynchronous in their cell cycles in vivo, making it difficult to discern differences in cell cycle regulation between the 2 cell types. Here, we treated C6G cells, a glioma cell line, and primary rat astrocytes with phase-specific cell-cycle inhibitors in vitro in order to provide a framework for understanding differences in the cell cycles of neoplastic and non-neoplastic astrocytes. By using both early removal and delayed addition of phase-specific inhibitors hydroxyurea and lovastatin to investigate the cell cycle kinetics in a glioma cell line (C6G cells) and in normal astrocytes, the present study further defines the differences in the cell cycle progression between these 2 cell types that were not characterized in our previous work.Citation25 Our current work reveals that C6G cells exhibit markedly different cell cycle kinetics as compared to primary astrocytes: they exhibit a shorter G1 phase and an earlier peak of S phase (), consistent with our previous findings.Citation25 In the present study, we also established an association between the DNA synthesis in C6G cells and 2 enzymes involved in cell cycle regulation of mammalian astrocytes – HMG-CoA reductase in late G1 phase, and ribonucleotide reductase in G1/S intersection and S phase. Our findings provide a basis on which to build future studies examining both the mechanisms of cell cycle dysregulation in tumoral astrocytes, as well as studies examining the efficacy of cell cycle inhibitors in treating gliomas in vivo.
The role of HMG-CoA reductase and its end product isoprenoids in modulating cell cycle progression through the restriction point in late G1 phase have been studied in different cell lines.Citation16,27-30 In mammalian astrocytes, biochemical abnormalities in HMG-CoA reductase have further been established in glioma cell lines.Citation21,22 The anti-proliferative and apoptotic effects of the HMG-CoA reductase inhibitor lovastatin on C6G cells were also characterized.Citation30 The mechanisms involved in the increased sensitivity of C6G cells to lovastatin remain unknown but might be related to biochemical abnormalities or increased expression of HMG-CoA reductase. Additional studies will be necessary to clarify this point.
In contrast to their sensitivity to lovastatin, C6G cells were resistant to hydroxyurea compared to primary astrocytes. The mainstay of chemotherapeutic agents for astrocytic neoplasms includes methylating agent temozolomide, as well as alkylating agents.Citation31,32 Survival rates associated with astrocytic neoplasms remain low as a result of the low efficacy of chemotherapy,Citation33 suggesting that glioma cells have efficient DNA repair mechanisms. Multiple and complicated molecular mechanisms of drug resistance for chemotherapeutic agents have been identified in different cancer cell lines.Citation34 These include mutation and amplification of target genes and involvement of the salvage pathway of nucleotide biosynthesis.Citation35-38 In several cell lines, resistance to hydroxyurea has been shown to involve gene amplification of the non-heme iron subunit (NHI) of ribonucleotide reductase, the target site of hydroxyurea.Citation34,36,39,40 Future studies will be necessary to unravel the mechanisms that underlie hydroxyurea resistance in C6 cells and other gliomas.
In the studies of delayed addition of lovastatin, a gradual loss of maximum inhibition was observed in primary astrocytes, but an abrupt decrease of 80% occurred by the 3 hour time point in C6G cells. Therefore, the IC50 in C6G cells occurred at a much earlier time point than in primary astrocytes (8 hours). This “leftward shift” of the IC50 in C6G cells is likely related to their early entry into S phase, as previous studies have demonstrated a functional role for HMG-CoA reductase in the restriction point, which is located in late G1 phase of mammalian astrocyte cell cycle.Citation1,16 On the other hand, the mechanisms involved in the gradual loss of inhibition in primary astrocytes during early G1 phase (3 hours and 6 hours) and the persistence of the inhibitory effect of lovastatin at or after G1/S intersection (12 hours and 15 hours) remain unknown and await further study. One possibility is that pathways other than the HMG-CoA reductase pathway are inhibited by lovasatatin, as seen in a breast cancer cell line.Citation41
Although C6G cells demonstrated greater sensitivity to the inhibitory effects of lovastatin on S phase DNA synthesis than primary astrocytes, they were also more likely to recover from a short exposure to lovastatin. Decreasing the incubation time of primary astrocytes by 18 hours (from 21 hours to 3 hours) only reduced the inhibition by 17%. On the other hand, a decrease in the exposure time by 9 hours (from 12 hours to 3 hours) in C6G cultures allowed C6G cells to recover from inhibition by 58%. A short 3-hour exposure to lovastatin resulted in a more significant inhibition in primary astrocytes than in C6G cells as a result of short exposure-induced “loss of inhibition” in C6G cells. This phenomenon is likely due to fact that HMG-CoA reductase, the rate-limiting enzyme in the biosynthesis of cholesterol and nonsterol products of mevalonate, is highly regulated by substrates and substrate-related products of the isoprenoids. Products of cholesterol biosynthetic pathways have also been shown to regulate the expression of HMG-CoA reductase.Citation42 Moreover, various inhibitors of HMG-CoA reductase, including lovastatin, induce its expression and activity.Citation43-46 This highly inducible and regulatory nature of HMG-CoA reductase has been established in several cell lines, but not in mammalian astrocytes or C6G cells.Citation42,44,46,47 Based on our data and that of others,Citation48 regulation of HMG-CoA reductase expression and activity may play an important role in the cell cycle progression of neoplastic astrocytes.
The “delayed addition and early removal” paradigm revealed that the addition of hydroxyurea immediately before G1/S intersection in both cell cultures (9 hours and 12 hours in primary astrocytes and 6 hours for C6G cells) resulted in much greater inhibition of DNA synthesis than addition at serum up-shift. Thereafter (during S phase), hydroxyurea progressively lost its inhibitory effects on DNA synthesis in both cell cultures. This “supernormal” inhibition was similar in both primary astrocytes and C6G cells, but was not seen when lovastatin was added in the same delayed fashion. This finding suggests that prolonged exposure to hydroxyurea prior to G1/S intersection will hamper hydroxyurea-mediated inhibition of DNA synthesis, as compared to addition of shortly before G1/S intersection. This “supernormal” inhibition was not likely due to a difference in membrane transport systems for hydroxyurea in these 2 cell types, as no transport system for hydroxyurea has been identified in mammalian cells.Citation49
In summary, our data reveal that C6G cells have more effective intrinsic mechanisms that enable them to recover from the inhibitory effects of hydroxyurea. Further studies will be needed to understand these mechanisms so as to maximize the efficacy of cancer chemotherapies.
Abbreviations
| 3-hydroxy-3-methylglutaryl coenzyme A | = | HMG-CoA |
| Bovine calf serum | = | BCS |
| Bromodeoxyuridine | = | BrDU |
| C6 glioma cells | = | C6G |
| Dulbecco's Modified Eagles medium | = | DMEM |
| Fetal bovine serum | = | FBS |
| Fluorescence-activated cell sorting | = | FACS |
| Non-heme iron subunit | = | NHI |
| Phosphate buffered saline | = | PBS |
| Retinoblastoma | = | Rb |
Disclosure of potential conflicts of interest
RC is a paid consultant on Mallinckrodt Pharmaceuticals' advisory boards. His role on the advisory boards is unrelated to the present studies. All other authors declare no competing interests.
Author contributions
Experiments were conceived and designed by VL, TJL, and RCC. Experiments were performed by VL and RCC. Data was analyzed by VL and RCC. Reagents/materials/tools provided by TJL. Manuscript was written and prepared by KRR and RCC. All authors reviewed and approved the final version of the manuscript.
Funding
The authors received no specific funding to perform these studies.
References
- Langan TJ, Slater MC. Cell cycling of astrocytes and their precursors in primary cultures: a mevalonate requirement identified in late G1, but before the G1/S transition, involves polypeptides. J Neurochem 1991; 56:1058-68; PMID:1993888; http://dx.doi.org/10.1111/j.1471-4159.1991.tb02029.x
- Langan TJ. Deterministic role of the cell cycle in newborn brain development and in brain injury. Semin Neurol 1993; 13:92-9; PMID:8511424; http://dx.doi.org/10.1055/s-2008-1041112
- Langan TJ, Slater MC. Astrocytes derived from long-term primary cultures recapitulate features of astrogliosis as they re-enter the cell division cycle. Brain Res 1992; 577:200-9; PMID:1606495; http://dx.doi.org/10.1016/0006-8993(92)90275-E
- Amat JA, Ishiguro H, Nakamura K, Norton WT. Phenotypic diversity and kinetics of proliferating microglia and astrocytes following cortical stab wounds. Glia 1996; 16:368-82; PMID:8721677; http://dx.doi.org/10.1002/(SICI)1098-1136(199604)16:4%3c368::AID-GLIA9%3e3.0.CO;2-W
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci USA 2005; 102:8333-8; PMID:15923260; http://dx.doi.org/10.1073/pnas.0500989102
- Hulleman E, Helin K. Molecular mechanisms in gliomagenesis. Adv Cancer Res 2005; 94:1-27; PMID:16095998; http://dx.doi.org/10.1016/S0065-230X(05)94001-3
- Cohen AL, Colman H. Glioma biology and molecular markers. Cancer Treat Res 2015; 163:15-30; PMID:25468223; http://dx.doi.org/10.1007/978-3-319-12048-5_2
- Li W, Li K, Zhao L, Zou H. Bioinformatics analysis reveals disturbance mechanism of MAPK signaling pathway and cell cycle in Glioblastoma multiforme. Gene 2014; 547:346-50; PMID:24967941; http://dx.doi.org/10.1016/j.gene.2014.06.042
- Koyama-Nasu R, Nasu-Nishimura Y, Todo T, Ino Y, Saito N, Aburatani H, Funato K, Echizen K, Sugano H, Haruta R, et al. The critical role of cyclin D2 in cell cycle progression and tumorigenicity of glioblastoma stem cells. Oncogene 2013; 32:3840-5; PMID:22964630; http://dx.doi.org/10.1038/onc.2012.399
- Saggioro FP, Neder L, Stávale JN, Paixão-Becker AN, Malheiros SM, Soares FA, Pittella JE, Matias CC, Colli BO, Carlotti CG Jr, et al. Fas, FasL, and cleaved caspases 8 and 3 in glioblastomas: a tissue microarray-based study. Pathol Res Pract 2014; 210:267-73; PMID:24561085; http://dx.doi.org/10.1016/j.prp.2013.12.012
- Ostrom QT, Gittleman H, Stetson L, Virk SM, Barnholtz-Sloan JS. Epidemiology of gliomas. Cancer Treat Res 2015; 163:1-14; PMID:25468222; http://dx.doi.org/10.1007/978-3-319-12048-5_1
- Dey M, Lin Y, Melkonian S, Lam S. Prognostic factors and survival in primary adult high grade brainstem astrocytoma: a population based study from 1973-2008. J Clin Neurosci 2014; 21:1298-303; PMID:24674698; http://dx.doi.org/10.1016/j.jocn.2013.12.011
- Bambury RM, Morris PG. The search for novel therapeutic strategies in the treatment of recurrent glioblastoma multiforme. Expert Rev Anticancer Ther 2014; 14:955-64; PMID:24814143; http://dx.doi.org/10.1586/14737140.2014.916214
- Giacinti C, Giordano A. RB and cell cycle progression. Oncogene 2006; 25:5220-7; PMID:16936740; http://dx.doi.org/10.1038/sj.onc.1209615
- Karlsson-Rosenthal C, Millar JB. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol 2006; 16:285-92; PMID:16682204; http://dx.doi.org/10.1016/j.tcb.2006.04.002
- Langan TJ, Iimori Y, White G, Volpe JJ. Regulation of sterol synthesis and of 3-hydroxy-3-methylglutaryl coenzyme A reductase by lipoproteins in glial cells in primary culture. J Neurosci Res 1987; 17:361-6; PMID:2887663; http://dx.doi.org/10.1002/jnr.490170406
- Langan TJ, Volpe JJ. Cell cycle-specific requirement for mevalonate, but not for cholesterol, for DNA synthesis in glial primary cultures. J Neurochem 1987; 49:513-21; PMID:3648095; http://dx.doi.org/10.1111/j.1471-4159.1987.tb02894.x
- Crick DC, Andres DA, Danesi R, Macchia M, Waechter CJ. Geranylgeraniol overcomes the block of cell proliferation by lovastatin in C6 glioma cells. J Neurochem 1998; 70:2397-405; PMID:9603204; http://dx.doi.org/10.1046/j.1471-4159.1998.70062397.x
- Quesney-Huneeus V, Wiley MH, Siperstein MD. Essential role for mevalonate synthesis in DNA replication. Proc Natl Acad Sci USA 1979; 76:5056-60; PMID:291922; http://dx.doi.org/10.1073/pnas.76.10.5056
- Mo H, Elson CE. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp Biol Med (Maywood) 2004; 229:567-85; PMID:15229351
- Volpe JJ, Obert KA. Coordinate regulation of cholesterol synthesis and 3-hydroxy-3-methylglutaryl coenzyme A synthase but not 3-hydroxy-3-methylglutaryl coenzyme A reductase in C-6 glia. Arch Biochem Biophys 1981; 212:88-97; PMID:6118099; http://dx.doi.org/10.1016/0003-9861(81)90346-5
- Maltese WA. Three-hydroxy-3-methylglutaryl coenzyme A reductase in human brain tumors. Neurology 1983; 33:1294-9; PMID:6684224; http://dx.doi.org/10.1212/WNL.33.10.1294
- Kikuchi T, Nagata Y, Abe T. In vitro and in vivo antiproliferative effects of simvastatin, an HMG-CoA reductase inhibitor, on human glioma cells. J Neurooncol 1997; 34:233-9; PMID:9258815; http://dx.doi.org/10.1023/A:1005753523949
- Murakami M, Goto T, Saito Y, Goto S, Kochi M, Ushio Y. The inhibitory effect of simvastatin on growth in malignant gliomas–with special reference to its local application with fibrin glue spray in vivo. Int J Oncol 2001; 19:525-31; PMID:11494031
- Li V, Kelly K, Schrot R, Langan TJ. Cell cycle kinetics and commitment in newborn, adult, and tumoral astrocytes. Brain Res Dev Brain Res 1996; 96:138-47; PMID:8922676; http://dx.doi.org/10.1016/0165-3806(96)00109-5
- Langan TJ, Chou RC. Synchronization of mammalian cell cultures by serum deprivation. Methods Mol Biol 2011; 761:75-83; PMID:21755442; http://dx.doi.org/10.1007/978-1-61779-182-6_5
- Larsson O, Zetterberg A. Effects of 25-hydroxycholesterol, cholesterol, and isoprenoid derivatives on the G1 progression in Swiss 3T3 cells. J Cell Physiol 1986; 129:94-102; PMID:3760035; http://dx.doi.org/10.1002/jcp.1041290114
- Larsson O. Role of biosynthesis of cholesterol and isoprenoid derivatives in regulation of G1 progression and cell proliferation of 3T6 cells. J Cell Physiol 1987; 133:163-8; PMID:3667703; http://dx.doi.org/10.1002/jcp.1041330121
- Larsson O, Wejde J. Dolichol delays G1-arrest for one cell cycle in human fibroblasts subjected to depletion of serum or mevalonate. J Cell Sci 1992; 103:1065-72; PMID:1487489
- Choi JW, Jung SE. Lovastatin-induced proliferation inhibition and apoptosis in C6 glial cells. J Pharmacol Exp Ther 1999; 289:572-9; PMID:10087052
- Gonzalez J, Gilbert MR. Treatment of astrocytomas. Curr Opin Neurol 2005; 18:632-8; PMID:16280673; http://dx.doi.org/10.1097/01.wco.0000191510.14627.d2
- Reardon DA, Rich JN, Friedman HS, Bigner DD. Recent advances in the treatment of malignant astrocytoma. J Clin Oncol 2006; 24:1253-65; PMID:16525180; http://dx.doi.org/10.1200/JCO.2005.04.5302
- Mathieu D, Fortin D. The role of chemotherapy in the treatment of malignant astrocytomas. Can J Neurol Sci 2006; 33:127-40; PMID:16736721; http://dx.doi.org/10.1017/S0317167100004881
- Cory AH, He AW, Cory JG. Multifactorial mechanisms of drug resistance in tumor cell populations selected for resistance to specific chemotherapeutic agents. Adv Enzyme Regul 1998; 38:3-18; PMID:9762343; http://dx.doi.org/10.1016/S0065-2571(97)00003-4
- Wettergren Y, Kullberg A, Levan G. Drug-specific rearrangements of chromosome 12 in hydroxyurea-resistant mouse SEWA cells: support for chromosomal breakage model of gene amplification. Somat Cell Mol Genet 1994; 20:267-85; PMID:7974003; http://dx.doi.org/10.1007/BF02254717
- Zhou BS, Hsu NY, Pan BC, Doroshow JH, Yen Y. Overexpression of ribonucleotide reductase in transfected human KB cells increases their resistance to hydroxyurea: M2 but not M1 is sufficient to increase resistance to hydroxyurea in transfected cells. Cancer Res 1995; 55:1328-33; PMID:7882331
- Levenson VV, Davidovich IA, Roninson IB. Pleiotropic resistance to DNA-interactive drugs is associated with increased expression of genes involved in DNA replication, repair, and stress response. Cancer Res 2000; 60:5027-30; PMID:11016623
- Kinsella AR, Smith D, Pickard M. Resistance to chemotherapeutic antimetabolites: a function of salvage pathway involvement and cellular response to DNA damage. Br J Cancer 1997; 75:935-45; PMID:9083327; http://dx.doi.org/10.1038/bjc.1997.164
- Carter GL, Thompson DP, Cory JG. Mechanisms of drug resistance to inhibitors directed at the individual subunits of ribonucleotide reductase. Cancer Commun 1989; 1:13-20; PMID:2701080
- Johnson CE, Hughes K, Cory JG. Cell-cycle associated transcriptional regulation of ribonucleotide reductase in L1210 leukemia cells and drug-resistant variants. Cancer Commun 1991; 3:341-9; PMID:1760249
- Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci USA 1999; 96:7797-7802; PMID:10393901; http://dx.doi.org/10.1073/pnas.96.14.7797
- Petras SF, Lindsey S, Harwood HJ Jr. HMG-CoA reductase regulation: use of structurally diverse first half-reaction squalene synthetase inhibitors to characterize the site of mevalonate-derived nonsterol regulator production in cultured IM-9 cells. J Lipid Res 1999; 40:24-38; PMID:9869647
- Bergstrom JD, Bostedor RG, Rew DJ, Geissler WM, Wright SD, Chao YS. Hepatic responses to inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase: a comparison of atorvastatin and simvastatin. Biochim Biophys Acta 1998; 1389:213-21; PMID:9512650; http://dx.doi.org/10.1016/S0005-2760(97)00182-3
- Li AC, Tanaka RD, Callaway K, Fogelman AM, Edwards PA. Localization of 3-hydroxy-3-methylglutaryl CoA reductase and 3-hydroxy-3-methylglutaryl CoA synthase in the rat liver and intestine is affected by cholestyramine and mevinolin. J Lipid Res 1988; 29:781-96; PMID:2902179
- Singer II, Scott S, Kazazis DM, Huff JW. Lovastatin, an inhibitor of cholesterol synthesis, induces hydroxymethylglutaryl-coenzyme A reductase directly on membranes of expanded smooth endoplasmic reticulum in rat hepatocytes. Proc Natl Acad Sci USA 1988; 85:5264-8; PMID:3293052 http://dx.doi.org/10.1073/pnas.85.14.5264
- Stone BG, Evans CD, Prigge WF, Duane WC, Gebhard RL. Lovastatin treatment inhibits sterol synthesis and induces HMG-CoA reductase activity in mononuclear leukocytes of normal subjects. J Lipid Res 1989; 30:1943-52; PMID:2621421
- Dawson PA, Metherall JE, Ridgway ND, Brown MS, Goldstein JL. Genetic distinction between sterol-mediated transcriptional and posttranscriptional control of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem 1991; 266:9128-34; PMID:1673968
- Rao KN. The significance of the cholesterol biosynthetic pathway in cell growth and carcinogenesis (review). Anticancer Res 1995; 15:309-14; PMID:7762999
- Morgan JS, Creasey DC, Wright JA. Evidence that the antitumor agent hydroxyurea enters mammalian cells by a diffusion mechanism. Biochem Biophys Res Commun 1986; 134:1254-9; PMID:3753869 http://dx.doi.org/10.1016/0006-291X(86)90385-2