
ABSTRACT
Polo-like kinases (PLKs) control several aspects of eukaryotic cell division and DNA damage response. Remarkably, PLKs are overexpressed in several types of cancer, being therefore a marker of bad prognosis. As such, specific PLK kinase activity inhibitors are already used in clinical trials and the regulation of PLK activation is a relevant topic of cancer research. Phosphorylation of threonine residues in the T-loop of the kinase domain is pivotal for PLKs activation. Here, we show that T238A substitution in the T-loop reduces the kinase activity of Cdc5, the only PLK in Saccharomyces cerevisiae, with minor effect on cell growth in unperturbed conditions. However, the cdc5-T238A cells have increased rate of chromosome loss and gross chromosomal rearrangements, indicating altered genome stability. Moreover, the T238A mutation affects timely localization of Cdc5 to the spindle pole bodies and blocks cell cycle restart after one irreparable double-strand break. In cells responding to alkylating agent metylmethane sulfonate (MMS), the cdc5-T238A mutation reduces the phosphorylation of Mus81-Mms4 resolvase and exacerbates the MMS sensitivity of sgs1Δ cells that accumulate Holliday junctions. Of importance, the previously described checkpoint adaptation defective allele, cdc5-ad does not show reduced kinase activity, defective Mms4 phosphorylation and genetic interaction with sgs1Δ. Our data define the importance of regulating Cdc5 activity through T-loop phosphorylation to preserve genome integrity and respond to DNA damage.
Introduction
Polo kinases (PLKs) are highly conserved mitotic regulators from yeast to mammals. Their number varies from just a single member in budding and fission yeast (Cdc5 & Plo1 respectively) to 5 members in mammals (PLK1-5). In all the eukaryotes, PLKs govern mitotic transition and cytokinesis, phosphorylating a number of different targets.Citation1 Importantly, balance of PLK1 level is very critical for normal cell cycle and genome stability as its overexpression is associated with various cancers, whereas its depletion induces aneuploidy.Citation2,3 In different organisms, PLKs were implicated in response to DNA damage to inactivate the DNA damage checkpoint (DDC).Citation4-6 More specifically, PLKs were involved in DDC inactivation and cell cycle restart either when DNA damage is repaired, thorough a process called checkpoint recovery, or when the DNA lesions are refractory to be repaired, through a process called checkpoint adaptation. Remarkably, checkpoint adaptation has been observed in higher eukaryotes and human cells responding to ionizing radiations and pharmacological concentration of various genotoxic agents. Also it has been postulated to drive tumorigenesis and resistance to oncotherapy.Citation7,8 Indeed, studies in yeast have reported that checkpoint adaptation precedes different types of genome instabilities.Citation9
It is now clear that in yeast and human cells, Cdc5 and PLK1 phosphorylate many factors involved in DNA damage checkpoint and repair. Importantly, they act directly on the checkpoint transducer kinases Rad53 and Chk2, inactivating the DDC.Citation10-14 In yeast, it is known that DDC activation restrains Cdc5 activity through phosphorylation, and recently it was shown that the protein is nuclearized.Citation15-18 Interestingly during checkpoint adaptation, Cdc5 is re-activated to inactivate critical mitotic regulators, such as Cdh1 and Bfa1, promoting spindle elongation and mitotic exit.Citation17-19
Considering their central role in many aspects of the DNA damage response and cell cycle progression, PLKs are finely regulated by different mechanisms.Citation20-22 All the PLKs are regulated through phosphorylation of Threonine residues in the T-loop of the kinase domain. Human PLK1 is phosphorylated at T210 in its activation loop by Aurora A and Aurora B kinases. The phosphorylation at T210 of human PLK1 by Aurora A with co-factor Bora is essential for early activation of the protein at centrosomes and also for checkpoint recovery.Citation23-26 In budding yeast, the T238 residue in the Cdc5 T-loop, which corresponds to T210 of PLK1, has also been found to be phosphorylated, but was shown to be dispensable for cell viability in unperturbed conditions.Citation27-30 It was also shown that the Cdc5 activity is primed by the CDK1 (Cdc28)-dependent phosphorylation of T242 in the T-loop of the kinase domain.Citation27,31
In this study, we further characterized the importance to phosphorylate the T238 residue in the T-loop of Cdc5 in S. cerevisiae. We found that this phosphorylation contributes to fully activate Cdc5 kinase activity, without grossly affecting cell growth in unperturbed conditions. Importantly, our results indicate that the phosphorylation of Cdc5 at T238 residue becomes critical for the cell to deal with DNA damage and preserve genome integrity.
Results
Phosphorylation at Thr238 of Cdc5 is dispensable for viability but its absence reduces the kinase activity of the protein
In S. cerevisiae, the activation loop of the kinase domain of Cdc5 is phosphorylated at 2 sites namely T238 and T242, which are conserved in higher eukaryotes (). Of note, phosphorylation of the T242 by Cdc28/CDK1 is required for the activation of Cdc5 and viability of the cell, whereas the functional role of the T238 phosphorylation has not been investigated in details.Citation27 Interestingly, the phosphorylation of equivalent site of the Cdc5-T238 in different orthologs was described to be important to fully activate Polo kinase.Citation20 Moreover, phosphorylation of the same site in PLK1 by Aurora A has been involved in DNA damage checkpoint recovery in human cells.Citation24,25 Thus, we decided to further investigate the role of T238 phosphorylation in Cdc5, focusing on DNA damage response and genome stability maintenance.
Figure 1. Phosphorylation of T238 site of Cdc5 is dispensable for cell viability. (A) An alignment of activation segment loops (T-loops) in diverse polo-kinases, showing the conserved T238, T242 and L251 residues of the ScCdc5. (B) Viability of the strains Y505 (cdc5-1), Y1327 (cdc5-1<empty vector>), Y1329 (cdc5-1 <CDC5::3xHA>), Y1331 (cdc5-1 <CDC5-N209A::3xHA>), Y1333 (cdc5-1 <CDC5-T238A::3xHA>) and Y1461 (cdc5-1 <CDC5-T242A::3xHA>), at different temperatures. Exponentially growing cell cultures were serially diluted (1:10), and each dilution was spotted on SC-Trp plates. Plates were incubated 3 d at the indicated temperatures.
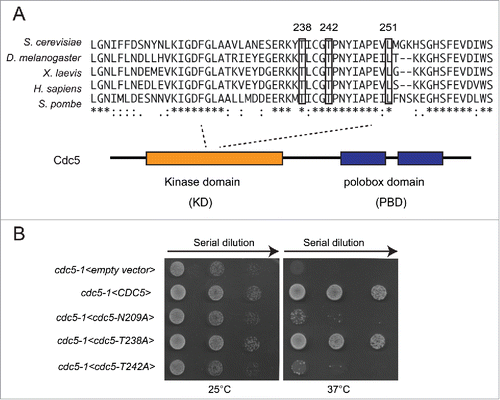
Firstly, we mutagenized the T238 or T242 sites to Alanine, a non-phosphorylable amino acid, in a plasmid carrying CDC5. Then, we analyzed the role of T242 and T238 phosphorylation in cell viability by assessing complementation of thermo-sensitive allele cdc5-1 at restrictive temperature. We also tested the wild type CDC5 and the kinase-dead cdc5-N209A alleles, as controls. As shown in , at non permissive temperature, the cells carrying thermo sensitive allele cdc5-1 are inviable due to failure to complete mitotic transition.Citation32 The thermo-sensitivity was completely rescued by expressing either the wild type CDC5 or the cdc5-T238A alleles on the plasmid. The expression of the kinase-dead cdc5-N209A and cdc5-T242A alleles did not rescue the cell lethality of cdc5-1 at 37°C, as described previously.Citation27 Therefore, our complementation assay supported previous finding,Citation27 indicating that the phosphorylation of the T238 in the T-loop of Cdc5 is dispensable for cell viability, while the phosphorylation of T242 by CDK1 is essential.
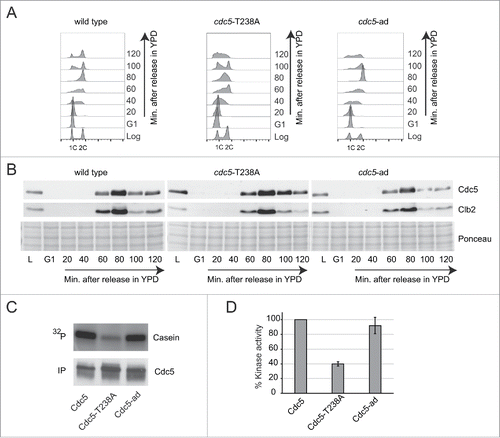
To further address the effect of the T238A mutation on Cdc5 protein level and kinase activity, as well as to investigate any potential impact on cell cycle and genome integrity, we integrated the cdc5-T238A allele at its endogenous locus. We compared cdc5-T238A mutant to the wild type and to another interesting allele, the cdc5-L251W (here after called cdc5-ad), which blocks DNA damage checkpoint adaptation.Citation33 As seen by cell plating and incubation at different temperatures, cdc5-T238A and cdc5-ad mutations did not significantly affect cell proliferation in unperturbed condition (Fig. S1A). However, a more accurate analysis by FACS performed on cells synchronized in G1 with α-Factor and released into fresh medium, showed that cdc5-T238A cells had a slight delay in late S and G2 phases (), suggesting dis-functions at these stages of the cell cycle. Nevertheless, both the Cdc5-T238A and Cdc5-ad protein variants were expressed throughout the cell cycle similarly to the wild type protein (). In particular, we observed that Cdc5-T238A and Cdc5-ad variants, as well as the wild type protein, were detectable in late S and G2/M phases, but not in late G1 and early S, mirroring the expression of the CDK1-cyclin Clb2. Indeed, it is known that Cdc5 is rapidly degraded in late G1, while it is stabilized and fully active in G2/M phase, when it is phosphorylated by CDK1-Clbs complex.Citation34,35 Then, we investigated Cdc5 protein level and stability in G1 and G2/M cell cycle phases in more details. Cells were kept blocked in G1 or G2/M with α-Factor or nocodazole treatment respectively, and protein samples were collected at indicated time points (Fig. S1B–C). As expected from the results in , the wild type Cdc5 protein and both the -T238A and -ad protein variants were expressed well in G2/M arrested cells, while they were rapidly degraded in G1 arrested cells (Fig. S1B–C). Then, we immunoprecipitated Cdc5 protein from the G2/M blocked cells to test in vitro its kinase activity, using Casein as substrate and γ-32P-ATP.Citation34 Surprisingly, we found that the Cdc5-T238A variant had almost 60% reduction in its kinase activity, compared to wild type protein (), while the Cdc5-ad retained the wild type level of kinase activity, as previously shown.Citation34 Supporting our observation, a recent study also highlights the role of T238 phosphorylation in regulating the kinase activity of Cdc5 in unperturbed conditions.Citation31
Figure 2. Expression and kinase activity analysis of the Cdc5-T238A and Cdc5-ad protein variants. (A) Cell cycle analysis by FACS of strains Y152 (CDC5::3xHA), Y1466 (cdc5-T238A::3xHA) and Y1465 (cdc5-ad::3xHA). Cells cultures were synchronized in G1 phase with α-Factor and released in fresh YPD medium. Samples for FACS analysis and protein extraction were collected at indicated time points. (B) Analysis of Cdc5 and Clb2 protein levels by western blot in same experiment described in (A). Blot stained with Ponceau is shown for gel loading control. (C-D) In vitro kinase assay performed with the indicated Cdc5 protein variants immunoprecipitated from Nocodazole-arrested cells (same strains as in A). The percent kinase activity of Cdc5-T238A and Cdc5-ad variants respect to the wild type Cdc5 are shown in a graph (D). Values are the mean of 3 independent experiments ± standard deviation.
cdc5-T238A cells do not adapt to one irreparable DSB and uncapped telomeres
Cdc5 has been found to promote checkpoint adaptation after one persistent DSB and telomere uncapping. In fact, cdc5-ad cells do not switch Rad53 off and do not re-start cell cycle after one irreparable HO-induced DSB.Citation33,36 We asked if cdc5-T238A cells have any effect on checkpoint inactivation after persistent DSB. We took advantage of yeast genetic background JKM139, in which an irreparable DSB is induced at MAT locus by the conditional over-expression of HO.Citation37 This is an ideal system to monitor checkpoint signaling and cell cycle progression, as it is unaffected by repair intermediates due to lack of homology sequences.Citation37,38 Thus, G1 unbudded cells were micro-manipulated in galactose containing medium to induce the HO-break. After DSB induction, activation of the DNA damage checkpoint blocks cell cycle progression at the G2/M transition for several hours.Citation38 However, wild type cells are known to undergo checkpoint adaptation, proceeding through 3–4 divisions after 24 hours, and are scored as the percent of cells forming micro-colonies. Strikingly, the number of cells that underwent adaptation was severely reduced in cdc5-T238A mutant similarly to the previously characterized cdc5-ad ( andCitation33). Of note, the cells with phospho-mimicking mutant cdc5-T238D were able to adapt proficiently (), further suggesting the hypothesis that the phosphorylation of T238 site of Cdc5 is a prerequisite for Cdc5 activity during checkpoint adaptation. Moreover, we repeated the assay using an untagged version of both the wild type and the cdc5-T238A strains, to rule out the possibility that the checkpoint adaptation defect might be due to a synthetic effect resulting from the presence of the 3xHA tag and the T238A mutation. Supporting previous observation in , we found that similar percentage of the wild type and cdc5-T238A cells underwent checkpoint adaptation at 24 hours after the formation of one irreparable HO-cut (Fig. S2A). Thus, C-terminal -3xHA tag does not affect the functionality of Cdc5 protein during checkpoint adaptation.
Figure 3. cdc5-T238A cells are defective in checkpoint adaptation to persistent DNA damage. (A) Graph shows the percentage of microcolonies with 3 or more cells plus buds for each mutant 24 hours after plating on galactose containing medium to induce one irreparable HO cut. Values are the mean of 3 independent experiments ± standard deviation. Representative images of cells/microcolonies for each strains 24 hours after plating are shown. The strains used are: Y152 (CDC5::3xHA), Y1466 (cdc5-T238A::3xHA), Y1465 (cdc5-ad::3xHA), and Y1573 (cdc5-T238D::3xHA). (B) Rad53 phosphorylation analysis by western blot of same strains as in (A) after one persistent HO-cut. (C-D) Analysis of nuclear division (DAPI) and spindle elongation (visualized with anti-α tubulin) after one persistent HO-cut analyzed in strains Y152 (CDC5::3xHA), Y1466 (cdc5-T238A::3xHA) and Y1465 (cdc5-ad::3xHA). (C) Representative images of cells after the indicated time after one irreparable HO-cut induction. The graph in (D) shows frequency of the indicated subpopulations at given time point as mean of 3 independent experiments ± standard deviation. Values were obtained counting at least 100 cells at each time point for each strain. (E) Graph shows the percentage of microcolonies with 3 or more cells plus buds for each strain 24 hours after plating on galactose containing medium to induce one irreparable HO cut. The strains used are: Y152 (CDC5::3xHA), Y1466 (cdc5-T238A::3xHA), Y1535 (cdc5-T238A::3xHA, rad9Δ), Y1574 (cdc5-T238A::3xHA, mad2Δ), Y3535 (cdc5-T238A::3xHA, bfa1Δ) and Y2155 (cdc5-T238A::3xHA, tel1Δ). Values are the mean of 3 independent experiments ± standard deviation.
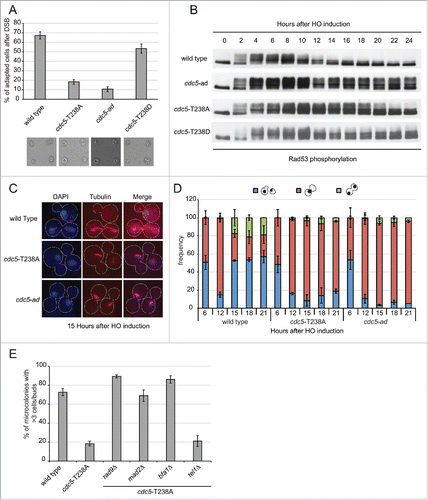
To address checkpoint adaptation at the molecular level in cdc5-T238A cells, we analyzed Rad53 phosphorylation by western blotting, after the induction of one HO-induced DSB. In wild type cells Rad53 is dephosphorylated after 12–15 hours after DSB induction ( andCitation36). In contrast, Rad53 dephosphorylation was severely impaired in cdc5-T238A cells till almost 20–22 hours, although the defect is less severe than in cdc5-ad cells (). In particular, we noted that in cdc5-T238A cells the percentage of cells adapting to irreparable DSB is very low, although Rad53 was significantly dephosphorylated at later time points. To further investigate this phenomenon, we monitored nuclear division accompanied by spindle elongation during checkpoint adaptation. Upon the induction of one irreparable DSB in logarithmically growing cells, the wild type cells switched-off checkpoint after 12–14 hours and underwent nuclear division accompanied by spindle elongation (). Consistently with the defect in checkpoint adaptation and micro-colony formation, both the cdc5-T238A and cdc5-ad cells remained blocked in metaphase with undivided nuclei at the bud neck and short spindle ().
Then, we combined cdc5-T238A mutation with some strains that carry deletion of the critical checkpoint genes TEL1, RAD9, MAD2, or the mitotic exit inhibitor BFA1. Indeed, it was previously shown that the maintenance of prolonged cell cycle block after the formation of one persistent DSB is mediated by the contributions of the spindle assembly checkpoint factor Mad2, in addition to the Rad53 activity.Citation39 Moreover, it was also shown that Rad53 dependent inhibition of Cdc5 in G2/M phase keeps Bfa1 in active state, thereby restricting mitotic spindle elongation and mitotic exit,Citation17 thus reinforcing the arrest. In addition, deletion of TEL1 is known to suppress a number of adaptation defective mutants (mec1-ad, sae2Δ, sgs1Δ and dna2Δ), which also had defects in DSB resection.Citation40,41
After micro-manipulating the cells in the presence of galactose to induce the HO-mediated irreparable DSB, we found that the permanent cell cycle block of cdc5-T238A cells was bypassed by deleting RAD9, MAD2 or BFA1 (), resembling what previously shown for cdc5-ad (Fig. S2B; andCitation39). Moreover, deletion of TEL1 did not overcome the cdc5-T238A mutant (), suggesting that this allele does not alter the initial processing of DSB ends.Citation40,41 For a more accurate analysis, we also repeated the irreparable HO-break response assay in the plate and counted the number of cells/microcolony in a time-course analysis (Fig. S3). The results indicated that the deletion of RAD9 lead to a complete bypass of the checkpoint arrest, whereas deletion of MAD2 and BFA1 did not completely abrogate the initial checkpoint arrest in cdc5-T238A cells, which in fact delayed at the G2/M transition for several hours before re-starting the cell cycle.
Collectively, the results in and Figure S3 suggest that the persistent cell cycle block after one irreparable DSB in cdc5-T238A cells might be caused by the inability to inactivate distinct targets, such as Rad53, Mad2 and Bfa1.
Of interest, we also found that cdc5-T238A cells were defective to re-start cell cycle after telomere uncapping in cdc13-1 background cells (Fig. S2C). Moreover, the persistent cell cycle block is bypassed by deleting RAD9. Thus, we conclude that cdc5-T238A mutation prevents checkpoint adaptation to uncapped telomeres, similarly to cdc5-ad.Citation33
Cdc5-T238A and Cdc5-ad protein variants show altered localization to spindle pole bodies after one irreparable DSB
Recent findings indicate that Cdc5 is localized into the nucleus after DNA damage and Rad53 activation,Citation17 thus preventing Bfa1 inactivation through Cdc5-dependent phosphorylation at Spindle Pole Bodies (SPBs). Based on these observations, we speculated that Cdc5 might relocalize at SPBs to inactivate Bfa1 and promote mitotic exit, during checkpoint adaptation. Thus, to analyze Cdc5 localization during checkpoint adaptation we inserted an eGFP tag to the C-terminal of Cdc5, Cdc5-T238A and Cdc5-ad proteins, in JKM139 background. After 6 hours of induction of one irreparable DSB, we observed that almost 80% of the cells got arrested in metaphase, with strong signal of Cdc5-eGFP in the nucleus. In wild type cells, after 10–12 hours of HO induction Cdc5 signal was specifically enriched at 2 perinuclear bright foci, corresponding to SPBs.Citation17,35 Consequently, greater number of cells with divided nuclei was observed during later time points (). Interestingly, even though the Cdc5-T238A protein variant was nuclearized after 6 hours of DSB induction, then we observed a prominent delay of its localization at SPBs at 16 – 18 hours (). This delay in Cdc5 localization may reflect the prolonged metaphase block with short spindle in cdc5-T238A cells, after one irreparable DSB. In the same experiment, we also investigated the localization of Cdc5-ad-eGFP protein variant. Surprisingly, we observed anticipated and persistent GFP signal at SPBs in cdc5-ad cells.
Figure 4. Analysis of Cdc5 localization during checkpoint adaptation. (A) Analysis of Cdc5-eGFP localization in CDC5-eGFP, cdc5-T238A-eGFP and cdc5-ad-eGFP strains in JKM139 background Y2228 (CDC5::eGFP), Y2230 (cdc5-ad::eGFP) and Y2232 (cdc5-T238A::eGFP). Galactose was added to induce one irreparable HO-cut in logarithmically growing cells. Samples were collected at indicated time points and observed for Cdc5-eGFP localization and nuclear morphology (DAPI). Representative images are shown. (B) Graph shows frequency of indicated subpopulations at given time point after HO induction. At least 100 cells were counted at each time point for each strain. Data represents mean of 3 independent experiments ± standard deviation.
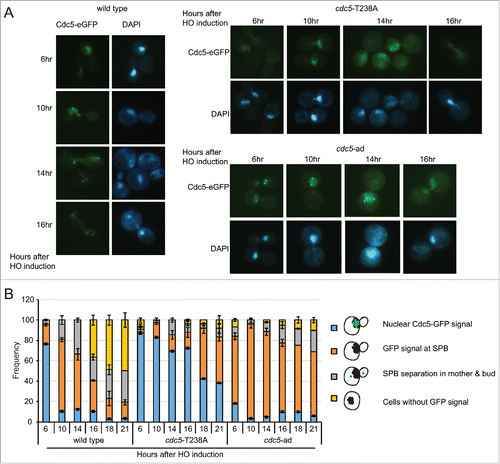
Altogether, our microscopic observations in indicate that both the Cdc5-T238A and Cdc5-ad protein variants localize to SPBs after one irreparable DSB with altered kinetic respect to the wild type protein. Intriguingly, our analysis suggest that misregulation at SPBs may contribute to prevent cell cycle re-start by checkpoint adaptation in cdc5-T238A and cdc5-ad cells, as recently shown in a separate class of cdc5 alleles, carrying mutations in the polo box domain.Citation42
cdc5-T238A affects the Mus81-Mms4 complex
Besides its role in checkpoint adaptation, Cdc5 mediates the phosphorylation and regulation of several proteins implicated in other aspects of the DNA damage response. One relevant Cdc5 target is Mms4, a regulatory subunit of the structure specific Mus81 endonuclease. Interestingly, we have recently shown that Mus81 is not involved in checkpoint adaptation after one persistent DSB.Citation43 Upon Cdc5-mediated activation, Mus81-Mms4 complex processes Holliday junctions (HJs), contributing to repair DNA lesions generated during stressful replication in the presence of genotoxic compounds, such as MMS.Citation44-47 In fact, the endonucleolytic activity mediated by the Mus81-Mms4 complex, together with the dissolution activity mediated by the Sgs1-Top3-Rmi1 complex (STR), represent the major pathways to process HJs (; andCitation48).
Figure 5. cdc5-T238A affects Mms4 phosphorylation and viability of sgs1Δ cells in MMS. (A) Model for processing of MMS-induced recombination intermediates through Sgs1-Top3-Rmi1 (STR) or Mus81-Mms4 complexes. (B) Mms4 phosphorylation analyzed by western blot. Cells cultures of the strains Y2147 (MMS4::3xHA), Y2206 (cdc5-T238A, MMS4::3xHA) and 2491 (cdc5-ad, MMS4::3xHA) were synchronised with α-Factor in G1 phase and released in YPD + nocodazole (noco). Samples were collected for FACS analysis and protein extraction at indicated time points. Blot stained with Ponceau is shown for gel loading control. (C) Same strains as in (B) were synchronised with α-Factor in G1 phase and released in YPD + MMS 0.02%. Samples were collected for FACS analysis and protein extraction at indicated time points. Blot stained with Ponceau is shown for gel loading control. Protein samples of wild type and cdc5-T238A strains treated with nocodazole for 2 hrs were loaded as control. (D) MMS sensitivity assay. Exponentially growing cell cultures of the strains: Y152 (CDC5::3xHA), Y1466 (cdc5-T238A::3xHA), Y1866 (sgs1Δ), Y1895 (cdc5-T238A::3xHA, sgs1Δ), Y1465 (cdc5-ad::3xHA), Y1893 (cdc5-ad::3xHA, sgs1Δ) were serially diluted (1:10), and each dilution was spotted out into YPD +/− the indicated dosages of MMS. Plates were incubated 3 d at 28°C.
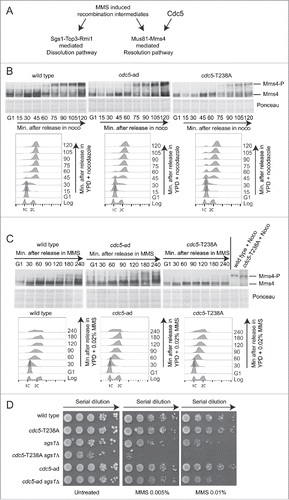
Investigating this regulatory network, we assessed Mms4 phosphorylation in cdc5-T238A and cdc5-ad cells. To this aim, we inserted a 3xHA tag at the C-terminal of Mms4 in wild type, cdc5-T238A and cdc5-ad strains. Cells were synchronized in G1 with α-Factor and released in fresh media containing nocodazole () or 0.02% MMS (). Samples were taken at the time points indicated in , and analyzed by FACS and western blotting. According to,Citation47 we observed a robust hyper-phosphorylation of Mms4 in wild type cells starting from 60–75 minutes after release in nocodazole (); a persistent MMS4 phosphorylation is also present from 120 minutes after the release in MMS (). Similar kinetic of Mms4 phosphorylation was observed in cdc5-ad cells, while it was severely delayed and lowered in cdc5-T238A cells, both in the presence of nocodazole and MMS (). This result is consistent with a Cdc5-dependent phosphorylation of Mms4,Citation44-47 and confirms our previous finding, indicating a reduced kinase activity of the Cdc5-T238A protein variant by in vitro assay (). Interestingly, when analyzed in nocodazole-blocked cells for 2 hours, the phosphorylation of Mms4 in cdc5-T238A cells did not look as much compromised than the wild type (), suggesting that the residual activity of Cdc5-T238A variant is sufficient for the Mms4 phosphorylation after a prolonged G2/M arrest.
Supporting that cdc5-T238A cells should retain reduced activity of the Mus81-Mms4 complex to process HJs, we hypothesized that cdc5-T238A mutation should increase the MMS sensitivity when introduced in sgs1Δ cells, which accumulate persistent dHJs.Citation49 Indeed MMS-induced HJs are primarily processed by the activity of the STR complex in S phase; then, persistent HJs are resolved later on by the activity of Mus81-Mms4.Citation50 Consistent with this, inactivating mutations in the STR and Mus81-Mms4 complexes are synthetic lethal.Citation51,52 Remarkably, the sgs1Δ cdc5-2 double mutant is viable, but exhibits increased chromosome missegregation and aneuploidy, as a result of improperly processed HJs.Citation45 Interestingly, we were able to generate both the sgs1Δ cdc5-T238A and sgs1Δ cdc5-ad double mutants. We found that the cdc5-T238A mutation, but not the cdc5-ad, is synthetic sick in combination with SGS1 deletion in unperturbed growth conditions and also exacerbates the severe sensitivity to MMS of the sgs1Δ cells (). This finding suggests that the reduced kinase activity of the Cdc5-T238A variant does not properly activate the Mus81-Mms4 complex, whose phosphorylation is reduced in cdc5-T238A cells (). As a consequence, the sgs1Δ cdc5-T238A double mutant cells should be severely defective in HJs processing and become extremely sensitive to MMS ().
cdc5-T238A mutation increases chromosome loss rate and gross chromosomal rearrangements
Based on its regulatory role of the DNA damage response, it is predicted that Cdc5 activity should be relevant to maintain genome integrity. Therefore, we investigated whether the cdc5-T238A mutation, apart from reducing the kinase activity of the protein (), may directly affect chromosome rearrangements and stability.
Firstly, we investigated chromosome loss rate in cdc5-T238A and cdc5-ad cells. To this aim, we used a modified genetic assay in which strain with stable Chromosome III fragment (CF) was created using CFV/D8B-tg as a result of break induced replication.Citation53 The presence of 110 kb CF was confirmed by Pulse-field gel electrophoresis. In W303 cells, the presence of SUP11 marker on CF suppresses the ade2-1 mutation leading to formation of white colonies, whereas the cells lacking the CF form red colonies (scheme in ). In this genetic background, the wild type cells have approximately ∼1 × 10−3 chromosome loss rate per cell per generation.Citation54 Interestingly, cdc5-T238A cells were found to increase the chromosome loss rate by 3-fold, while cdc5-ad cells were almost similar to wild type (). This result suggests that a fully active Cdc5 is important to coordinate the events to maintain stable chromosomes, likely affecting their replication and segregation. Moreover, the fact that the cdc5-ad mutation did not increase chromosome loss rate, may indicate that the fold increase found in the cdc5-T238A cells is likely related to the reduced kinase activity of Cdc5 variant than to the checkpoint-adaptation defect per se.
Figure 6. cdc5-T238A cells have altered genome stability. (A) Schematic representation of chromosome loss assay to determine the chromosome loss rate per cell per generation. Only half red-sectored events were scored (see materials and methods). (B) Graph showing chromosome loss rate of strains Y1973 (wild type) Y1979 (cdc5-T238A) and Y2371 (cdc5-ad). P value was calculated by 2 tailed student's t-test. The data represents at least 3 independent experiments. (C) Schematic representation of gross chromosomal rearrangement (GCR) assay. (D) GCR rate was measured in the strains: Y101C7/C8 (wild type), Y3359/60 (cdc5-T238A), Y3510/32 (sgs1Δ) and Y3529/30 (cdc5-T238A::3xHA, sgs1Δ) by selecting for simultaneous loss of the CAN1 and URA3 genes on chromosome V. The graph shows mean and standard deviations of at least 2 independent biological isolates for each mutant.
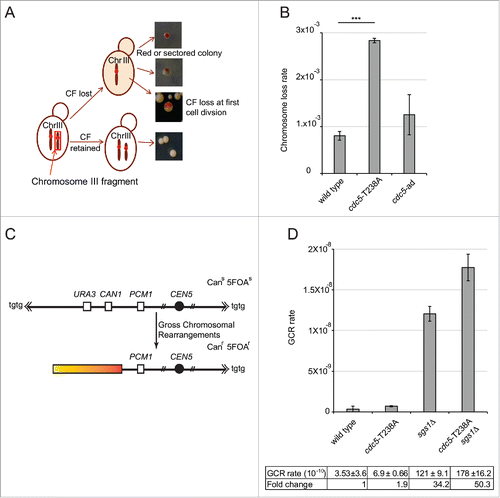
Then, to further address the impact on genome integrity of the cdc5-T238A mutation, we integrated the cdc5-T238A allele in a specific genetic background, commonly used to analyze gross chromosomal rearrangements (GCR).Citation55 In this genetic system, the URA3 and CAN1 genes on a non-essential arm of a specialized chromosome V confer sensitivity to 5-fluoroorotic acid (5-FOA) and canavanine. However, the 2 counter-selectable markers can be lost due to spontaneous breaks formation along the chromosome arm and subsequent GCR, which includes loss of chromosome arm and de-novo telomere addition, non-reciprocal translocations and chromosome fusions, or interstitial deletions (). Using this system, it was shown that several DNA repair and checkpoint genes are involved to suppress these events. Of importance, deletion of SGS1 leads to a dramatic increase of GCRs.Citation56,57 Considering the genetic interaction between sgs1Δ and cdc5-T238A mutations (), we tested GCRs in the sgs1Δ cdc5-T238A double mutant, together with the corresponding single mutants. We found that cdc5-T238A mutation resulted only in 2-fold increase in GCR that seen in wild type; however, surprisingly it elevated GCR events in sgs1Δ cells to a value significantly higher than observed in the corresponding single mutants (). These observations suggest that Cdc5 activity contributes to suppress GCRs through a Sgs1-independent pathway, giving a critical contribution in sgs1Δ cells. Indeed, Cdc5 may phosphorylate and regulate several factors that can suppress GCRs. Among these Cdc5 targets, one interesting candidate is the Mus81-Mms4 complex, which has already been involved in suppressing GCRs,Citation56 and it is less phosphorylated and active in cdc5-T238A cells ().
Taken together the results in , it becomes evident that the cdc5-T238A mutation affects chromosome stability and rearrangement, indicating an involvement of Cdc5 kinase activity in preserving genome integrity in unperturbed conditions.
Discussion
PLKs are activated through phosphorylation of well-conserved Threonine sites in the T-loop of the kinase domain.Citation20 A relevant example of this regulation was shown for the human PLK1, in which T210, the first Threonine residue of the T-loop motif, is phosphorylated by Aurora kinases in cooperation with Bora, activating PLK1 even after DNA damage.Citation24-26 Interestingly, the phosphorylation of the first T residue of the T-loop is critical for the activation of most PLKs expressed in different organisms.Citation20 The T238 site of Cdc5 in S. cerevisiae, corresponding to T210 of PLK1 (), has been found phosphorylated by an unknown kinase.Citation27,31 So far, the functional role of Cdc5 T238 phosphorylation has been poorly studied, because the cdc5-T238A mutation does not affect much cell viability in unperturbed cell cycle ( andCitation27). Moreover, it was shown that the Cdc28-dependent phosphorylation of T242, the second T residue in the T-loop of Cdc5, is mainly responsible of the full activation of the kinase domain and is essential for cell viability.Citation27,31
In this study, we further address the functional role of the phosphorylation of Cdc5 at the T238 site. We found that the Cdc5-T238A protein variant retains a significantly reduced kinase activity by in vitro assay, although cdc5-T238A cells grow almost as the wild type in unperturbed conditions ( and ). We only observed a slight delay in S/G2 (), which may be associated with defects in chromosome replication and/or segregation, as well as activation of the Mitotic Exit Network (MEN), that, however, do not compromise cell proliferation (see also below). Remarkably, cdc5-T238A cells are defective in checkpoint adaptation after inducing one irreparable DSB, and remain blocked in G2/M with prolonged Rad53 phosphorylation and short spindle (). This persistent checkpoint, even though inactivated at later time points in cdc5-T238A cells, is detrimental for the cells, which in fact do not restart cell division even after 24 hours ( and Fig. S3). These observations indicate that Cdc5 activity is necessary for efficient checkpoint adaptation not only to silence the DDC, but also to govern the network of events leading to the re-start of cell division after the DNA damage-induced delay. Of importance, this finding supports the notion that checkpoint adaptation should proceed through the optimized coordination of multiple events, dispelling the idea that only mutations that prevent DDC silencing exclusively affect the entire process. Strikingly, the permanent cell cycle block observed in cdc5-T238A cells after one irreparable DSB is bypassed either by deletion of RAD9, MAD2 or BFA1 ( and Fig. S3), suggesting that checkpoint adaptation and cell cycle re-start after DNA damage require fine interplay between Cdc5 and multiple factors and pathways. Indeed, recent studies indicated that Cdc5 is localized into the nucleus in presence of DNA damage and it is speculated that it should relocalize to SPBs to inactivate inhibitors of mitosis and cell cycle regulators, i.e. Bfa1-Bub2 complex, Mad2 (component of Spindle Assembly checkpoint) and Cdh1 (inhibitor of spindle elongation).Citation17-19 Moreover, a clear link between the localization of Cdc5 at centrosomes and checkpoint adaptation to persistent DNA damage and spindle depolarization has been recently established.Citation42,58 Of interest, we found that the Cdc5-T238A protein variant relocalizes to SPBs with several hours of delay, according to the prolonged cell cycle block after one irreparable DSB (). Possibly, this defect at SPBs may also affect chromosome segregation and stability. As such, we found modest, but reproducible increase in chromosome loss rate in cdc5-T238A (). Furthermore, cdc5-T238A mutation significantly elevates sensitivity to MMS and spontaneous GCRs in sgs1Δ cells ( and ). Working as DNA helicase of the STR complex, Sgs1 dissolves DNA joint molecules formed during HR and stressful replication. In its absence, recombination intermediates accumulate and have to be processed by Mus81-MMS4 and Yen1 structure-specific endonucleases.Citation48 Interestingly, we found reduced phosphorylation of Mms4 in cdc5-T238A cells (), likely explaining the genetic interaction of this allele with sgs1Δ. Indeed, a Cdc5-dependent phosphorylation of Mms4 was shown to activate the Mus81-Mms4 endonuclease complex to process HJs.Citation44-47 Therefore, we can speculate that unprocessed HJs accumulate in the sgs1Δ cdc5-T238A cells, leading to aberrant chromosome segregation and loss, increase GCRs and hyper-sensitivity to MMS. Most important, our results in on genome rearrangement and chromosome stability, taken together with the recently involvement of Cdc5 in maintaining genome ploidy,Citation42 further support Cdc5 as key factor to preserve genome integrity.
Interestingly, our results differentiate the cdc5-T238A allele from previously reported adaptation-defective cdc5-ad allele.Citation33 Indeed, contrary to the cdc5-T238A cells, the cdc5-ad cells do not show: i) reduced kinase activity by in vitro assay (, andCitation34); ii) reduced Mms4 phosphorylation (); iii) increased MMS sensitivity in sgs1Δ cells (); iv) increased chromosome loss (). Moreover, SPBs localization of Cdc5-ad variant is anticipated (), likely reflecting the frequent nuclear excursion already documented in cdc5-ad cells.Citation39,59 Thus, we believe that further characterization of both the alleles will be important to investigate Cdc5 role in genome integrity.
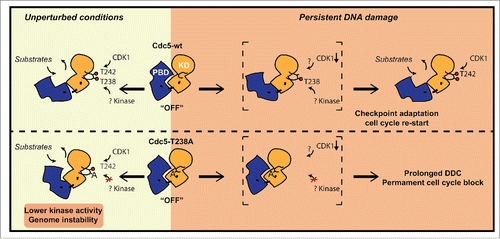
In summary, we show that the phosphorylation of T238 residue in the T-loop domain of Cdc5 contributes to fully activate Cdc5, controlling several events to preserve genome integrity and to adapt to a persistent DSB. At the molecular level, similarly to what has been shown for the regulation of PLK1,Citation21 we can speculate that the phosphorylation of the conserved T238 site may contribute to reduce the interaction between the kinase domain and the PBD, leading to the full activation of kinase domain and to the release of PBD for target interaction and centrosome localization. This mechanism can be particularly important to activate Cdc5 when the phosphorylation of the T242 site in the T-loop is compromised, such as when the Cdc28 activity is kept low in the presence of DNA damage (see a model in ). Contrary to cdc5-ad, which is thought to affect phosphorylation of a subset of Cdc5 substrates,Citation33 mutation T238A compromises full activation of the protein, thus precluding complete phosphorylation of a broad range of Cdc5 targets. As such, cdc5-T238A allele promises to be a useful reagent for global approaches to identify all the Cdc5 targets that are involved in DNA repair and genetic stability, with expected conservation in human cells.
Figure 7. Schematic model representing the regulation of Cdc5 activity through T238 phosphorylation in T-loop of kinase domain. Based on our results, we propose that T238 phosphorylation has important implications for maintaining genome stability in unperturbed conditions. Moreover it becomes essential for checkpoint adaptation and cell cycle restart after persistent DNA damage. Red circles indicate phosphorylation events. Abbreviations: KD (Kinase Domain); PBD (Polo Box Domain).
This study underlines an important role of T-loop phosphorylation of Polo kinase Cdc5 to maintain genome integrity and promote cell cycle re-entry after persistent DNA damage. Our results may be of interest to develop novel and specific PLK1 inhibitors to modulate its kinase activity for cancer treatment,Citation2,3 also aiming to reduce unwanted side effects and genome instability.
Materials and methods
Strains and growth conditions
All the strains listed in Table S1 are derivative of JKM139, BY4741 or W303. To construct strains standard genetic procedures of transformation and tetrad analysis were followed. Deletions and tag fusions were generated by the one-step PCR system.Citation60 Mutant alleles of CDC5 were obtained by site-specific mutagenesis of pRS306 plasmid containing wild type CDC5 with its endogenous promoter and C-terminal –HA tag. BclI-digested pRS306 plasmid was integrated into the CDC5 locus and after pop-out by treatment with 5-FOA; the integration of the cdc5-T238A and other alleles was confirmed by sequencing. Except the complementation analysis of cdc5-1 (), all the experiments were performed with CDC5 mutations integrated at its endogenous locus.
Strains used for chromosome loss assay were generated by transforming SnaBI digested CFV/D8B-tg into RAD5 derivative of W303 background. Stable Ura+ transformants due to BIR induced extra-chromosome fragment were confirmed by pulse field gel electrophoresis as described previously.Citation53 For the indicated experiments, cells were grown in YP medium either enriched with 2% glucose (YEP+glu, also indicated as YPD), raffinose 3% (YEP+raf) or raffinose 3% and galactose 2% (YEP+raf+gal). All the synchronization experiments were performed at 28°C.
Western blot analysis
The TCA protein extraction and the western blot procedures have been previously described.Citation61 Clb2, Rad53 and -3HA tagged proteins were analyzed using Clb2 (y-180, sc-9071) (Santa Cruz Biotech), Mab.EL7, and 12CA5 monoclonal antibodies, respectively.
Cell synchrony and flow cytometry
Cells were pre-synchronized in G1 with α-factor (2 µg/ml) and then released in fresh medium. Cells were arrested in G1 and G2/M with α-Factor (10 µg/ml) or nocodazole (20 µg/ml), respectively. DNA content was analyzed by FACS Calibur (Bekton-Dickinson) and Cell-Quest software (Bekton-Dickinson).
Immunofluorescence analysis
Samples were collected at indicated time points and fixed either in 100% ethanol or K-Phos.-formaldehyde with magnesium chloride buffer. Spheroplasting was done with 1mg/ml of zymoliase. Monoclonal anti-α tubulin antibody was used to visualize tubulin and nuclei were stained with DAPI. Images were captured using Leica BG DMR fluorescence microscope and analyzed with LAS AF suite.
In vitro kinase assay
Cdc5-3HA kinase activity was measured in 12CA5 immuno-precipitates from nocodazole arrested cells and washed sequentially in LLB, high-salt QA (20 mM Tris-HCl, pH 7.6, 250 mM KCl, 1 mM MgCl2, 1 mM DTT), and 5KB (50 mM Hepes-NaOH, pH 7.4, 200 mM KAc, 10 mM MgCl2, 5 mM MnCl2, 1 mM DTT). Kinase assays (30µl) were performed in 50 mM Hepes-NaOH, pH 7.4, 60 mM KAc, 10 mM MgCl2, 5 mM MnCl2, 50 mM ATP, plus 5 mg casein and 2.5 µCi [32P]ATP.Citation34
Checkpoint adaptation analysis by micro-colony assay
JKM139 derived strains were grown overnight in YP+raf media and 100 unbudded cells (G1 phase) for each strain were micro-manipulated on YEP+raf+gal plates. Percentage of checkpoint adaptation was scored after 24 and 48 hrs of incubation. For cdc13-1 derived strains, cells were grown overnight in YP+Glu at 23°C and were shifted to 37°C, 2 hours before micro-manipulation on YPD plates to induce telomere uncapping. After micromanipulation plates were incubated at 37°C for 24 hours.
Chromosome loss assay
Strains with chromosome III fragment (110kb) were grown overnight in SC-uracil liquid medium. The following day, cells were washed with sterile water and plated on SC+Ade (6µg/ml) to enhance red pigmentation. After incubation of 3–4 days, at least 10,000 colonies were screened per strain for exact half red/white sectoring which indicates chromosome loss at first cell division in non-selective medium.Citation54 The data represents 3 independent experiments.
Spot test for DNA damage sensitivities
Log phase cultures were normalized to 107cells/ml and 10µl of tenfold serial dilutions were spot plated on control and drug containing YPD plates. Plates were incubated at 28°C for 3 d.
Gross chromosomal rearrangement rate measurement
GCR were measured as described previously.Citation55 Briefly, 7 isolated colonies were inoculated in 40 ml of YPD and were grown till saturation for 3 d. Cultures were centrifuged and resuspended in sterile water (0.2ml/10 ml of culture) and ∼ 109 cells were plated on plates containing 1mg/ml FOA, 60µg/ml Canavanine. Simultaneously, serial dilutions of cultures were plated on YPD plates for determining viable cells in the culture. FOAr Canr colonies were measured after incubating the plates at 28°C for 4–5days. GCR was calculated using Lea-Coulson's method of median. Data represents rate of 2 independent biological isolates.
Abbreviations
| CDK | = | Cyclin dependent kinase |
| PLK | = | Polo-like kinase |
| PBD | = | Polo box domain |
| MMS | = | methyl methanesulphonate |
| DDC | = | DNA damage checkpoint |
| DSB | = | Double strand break |
| SPB | = | Spindle pole body |
| STR complex | = | Sgs1-Top3-Rmi1 complex |
| dHJ | = | DNA double Holliday junctions |
| GCR | = | Gross chromosomal rearrangements |
| MEN | = | Mitotic exit network. |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Figure_S1.eps
Download EPS Image (20.9 MB)Figure_S2.eps
Download EPS Image (1.4 MB)Figure_S3.eps
Download EPS Image (3.9 MB)Supplementary Files
Download PDF (111.1 KB)Acknowledgments
We thank Vladimir Botchkarev and James Haber for communicating results prior to publication, and for critically reading of the manuscript. We acknowledge J. E. Haber, L. S. Symington and G. W. Brown for generously providing yeast strains and plasmids. We are grateful to our lab members and G. Liberi for helpful discussions and comments.
Funding
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro [AIRC_IG Grant n.15488 to A.P; CARIPLO [2013–0790 to A.P.]; a fellowship from Fondazione “Gabriella Dolfin Voyasidis”-Accademia Nazionale dei Lincei to M.F. Funding for open access charge [Associazione Italiana per la Ricerca sul Cancro/ AIRC_IG Grant n.15488].
Related Research Data
References
- Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol 2009; 10:265-75; PMID:19305416; http://dx.doi.org/10.1038/nrm2653
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010; 9:643-60; PMID:20671765; http://dx.doi.org/10.1038/nrd3184
- Liu X. Targeting polo-like kinases: A promising therapeutic approach for cancer treatment. Trans Oncol 2015; 8:185-95; http://dx.doi.org/10.1016/j.tranon.2015.03.010
- Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19:238-45; PMID:17303408; http://dx.doi.org/10.1016/j.ceb.2007.02.009
- Serrano D, D'Amours D. When genome integrity and cell cycle decisions collide: roles of polo kinases in cellular adaptation to DNA damage. Syst Synth Biol 2014; 8:195-203; PMID:25136381; http://dx.doi.org/10.1007/s11693-014-9151-9
- Shaltiel IA, Krenning L, Bruinsma W, Medema RH. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci 2015; 128:607-20; PMID:25609713; http://dx.doi.org/10.1242/jcs.163766
- Swift LH, Golsteyn RM. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Inter J Mol Sci 2014; 15:3403-31; http://dx.doi.org/10.3390/ijms15033403
- Syljuasen RG, Jensen S, Bartek J, Lukas J. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res 2006; 66:10253-7; PMID:17079442; http://dx.doi.org/10.1158/0008-5472.CAN-06-2144
- Galgoczy DJ, Toczyski DP. Checkpoint adaptation precedes spontaneous and damage-induced genomic instability in yeast. Mol Cell Biol 2001; 21:1710-8; PMID:11238908; http://dx.doi.org/10.1128/MCB.21.5.1710-1718.2001
- Donnianni RA, Ferrari M, Lazzaro F, Clerici M, Tamilselvan Nachimuthu B, Plevani P, Muzi-Falconi M, Pellicioli A. Elevated levels of the polo kinase Cdc5 override the Mec1/ATR checkpoint in budding yeast by acting at different steps of the signaling pathway. PLoS Genet 2010; 6:e1000763; PMID:20098491; http://dx.doi.org/10.1371/journal.pgen.1000763
- Schleker T, Shimada K, Sack R, Pike BL, Gasser SM. Cell cycle-dependent phosphorylation of Rad53 kinase by Cdc5 and Cdc28 modulates checkpoint adaptation. Cell Cycle 2010; 9:350-63; PMID:20046099; http://dx.doi.org/10.4161/cc.9.2.10448
- Vidanes GM, Sweeney FD, Galicia S, Cheung S, Doyle JP, Durocher D, Toczyski DP. CDC5 inhibits the hyperphosphorylation of the checkpoint kinase Rad53, leading to checkpoint adaptation. PLoS Biol 2010; 8:e1000286; PMID:20126259; http://dx.doi.org/10.1371/journal.pbio.1000286
- van Vugt MA, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong SE, Tan CS, Miao H, Keezer SM, Li J, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol 2010; 8:e1000287; PMID:20126263; http://dx.doi.org/10.1371/journal.pbio.1000287
- Yoo HY, Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 2004; 117:575-88; PMID:15163406; http://dx.doi.org/10.1016/S0092-8674(04)00417-9
- Cheng L, Hunke L, Hardy CF. Cell cycle regulation of the Saccharomyces cerevisiae polo-like kinase cdc5p. Mol Cell Biol 1998; 18:7360-70; PMID:9819423; http://dx.doi.org/10.1128/MCB.18.12.7360
- Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 1999; 286:1166-71; PMID:10550056; http://dx.doi.org/10.1126/science.286.5442.1166
- Valerio-Santiago M, de Los Santos-Velazquez AI, Monje-Casas F. Inhibition of the mitotic exit network in response to damaged telomeres. PLoS Genet 2013; 9:e1003859; PMID:24130507; http://dx.doi.org/10.1371/journal.pgen.1003859
- Zhang T, Nirantar S, Lim HH, Sinha I, Surana U. DNA damage checkpoint maintains CDH1 in an active state to inhibit anaphase progression. Developmental cell 2009; 17:541-51; PMID:19853567; http://dx.doi.org/10.1016/j.devcel.2009.09.006
- Crasta K, Lim HH, Giddings TH, Jr., Winey M, Surana U. Inactivation of Cdh1 by synergistic action of Cdk1 and polo kinase is necessary for proper assembly of the mitotic spindle. Nat Cell Biol 2008; 10:665-75; PMID:18500339; http://dx.doi.org/10.1038/ncb1729
- Archambault V, Carmena M. Polo-like kinase-activating kinases: Aurora A, Aurora B and what else? Cell Cycle 2012; 11:1490-5; PMID:22433949; http://dx.doi.org/10.4161/cc.19724
- Archambault V, Lepine G, Kachaner D. Understanding the Polo Kinase machine. Oncogene 2015; 34:4799-807; PMID:25619835; http://dx.doi.org/10.1038/onc.2014.451
- Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 2004; 5:429-40; PMID:15173822; http://dx.doi.org/10.1038/nrm1401
- Bruinsma W, Aprelia M, Kool J, Macurek L, Lindqvist A, Medema RH. Spatial separation of Plk1 phosphorylation and activity. Front Oncol 2015; 5:132; PMID:26114094; http://dx.doi.org/10.3389/fonc.2015.00132
- Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455:119-23; PMID:18615013; http://dx.doi.org/10.1038/nature07185
- Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008; 320:1655-8; PMID:18566290; http://dx.doi.org/10.1126/science.1157425
- Tsvetkov L, Stern DF. Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage. Cell Cycle 2005; 4:166-71; PMID:15611664; http://dx.doi.org/10.4161/cc.4.1.1348
- Mortensen EM, Haas W, Gygi M, Gygi SP, Kellogg DR. Cdc28-dependent regulation of the Cdc5/Polo kinase. Curr Biol 2005; 15:2033-7; PMID:16303563; http://dx.doi.org/10.1016/j.cub.2005.10.046
- Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics 2008; 7:1389-96; PMID:18407956; http://dx.doi.org/10.1074/mcp.M700468-MCP200
- Jones MH, Keck JM, Wong CC, Xu T, Yates JR, 3rd, Winey M. Cell cycle phosphorylation of mitotic exit network (MEN) proteins. Cell Cycle 2011; 10:3435-40; PMID:22031224; http://dx.doi.org/10.4161/cc.10.20.17790
- Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villen J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Meth 2013; 10:676-82; http://dx.doi.org/10.1038/nmeth.2519
- Rodriguez-Rodriguez JA, Moyano Y, Jativa S, Queralt E. Mitotic Exit Function of Polo-like Kinase Cdc5 Is Dependent on Sequential Activation by Cdk1. Cell Rep 2016; PMID:27210759
- Hartwell LH, Mortimer RK, Culotti J, Culotti M. Genetic control of the cell division cycle in yeast: V. genetic analysis of cdc mutants. Genetics 1973; 74:267-86; PMID:17248617
- Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997; 90:1097-106; PMID:9323137; http://dx.doi.org/10.1016/S0092-8674(00)80375-X
- Charles JF, Jaspersen SL, Tinker-Kulberg RL, Hwang L, Szidon A, Morgan DO. The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr Biol 1998; 8:497-507; PMID:9560342; http://dx.doi.org/10.1016/S0960-9822(98)70201-5
- Shirayama M, Zachariae W, Ciosk R, Nasmyth K. The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. Embo J 1998; 17:1336-49; PMID:9482731; http://dx.doi.org/10.1093/emboj/17.5.1336
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell 2001; 7:293-300; PMID:11239458; http://dx.doi.org/10.1016/S1097-2765(01)00177-0
- White CI, Haber JE. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. Embo J 1990; 9:663-73; PMID:2178924
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 1998; 94:399-409; PMID:9708741; http://dx.doi.org/10.1016/S0092-8674(00)81482-8
- Dotiwala F, Harrison JC, Jain S, Sugawara N, Haber JE. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr Biol 2010; 20:328-32; PMID:20096585; http://dx.doi.org/10.1016/j.cub.2009.12.033
- Clerici M, Trovesi C, Galbiati A, Lucchini G, Longhese MP. Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. Embo J 2014; 33:198-216; PMID:24357557
- Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol 2012; 32:4727-40; PMID:23007155; http://dx.doi.org/10.1128/MCB.00566-12
- Ratsima H, Serrano D, Pascariu M, D'Amours D. Centrosome-dependent bypass of the DNA damage checkpoint by the Polo Kinase Cdc5. Cell reports 2016; 14:1422-34; PMID:26832404; http://dx.doi.org/10.1016/j.celrep.2016.01.014
- Dibitetto D, Ferrari M, Rawal CC, Balint A, Kim T, Zhang Z, Smolka MB, Brown GW, Marini F, Pellicioli A. Slx4 and Rtt107 control checkpoint signalling and DNA resection at double-strand breaks. Nucleic Acids Res 2016:669-82; PMID:26490958; http://dx.doi.org/10.1093/nar/gkv1080
- Matos J, Blanco MG, Maslen S, Skehel JM, West SC. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 2011; 147:158-72; PMID:21962513; http://dx.doi.org/10.1016/j.cell.2011.08.032
- Matos J, Blanco MG, West SC. Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep 2013; 4:76-86; PMID:23810555; http://dx.doi.org/10.1016/j.celrep.2013.05.039
- Schwartz EK, Wright WD, Ehmsen KT, Evans JE, Stahlberg H, Heyer WD. Mus81-Mms4 functions as a single heterodimer to cleave nicked intermediates in recombinational DNA repair. Mol Cell Biol 2012; 32:3065-80; PMID:22645308; http://dx.doi.org/10.1128/MCB.00547-12
- Gallo-Fernandez M, Saugar I, Ortiz-Bazan MA, Vazquez MV, Tercero JA. Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res 2012; 40:8325-35; PMID:22730299; http://dx.doi.org/10.1093/nar/gks599
- Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci 2014; 39:409-19; PMID:25131815; http://dx.doi.org/10.1016/j.tibs.2014.07.003
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 2005; 19:339-50; PMID:15687257; http://dx.doi.org/10.1101/gad.322605
- Szakal B, Branzei D. Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. Embo J 2013; 32:1155-67; PMID:23531881; http://dx.doi.org/10.1038/emboj.2013.67
- Fabre F, Chan A, Heyer WD, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A 2002; 99:16887-92; PMID:12475932; http://dx.doi.org/10.1073/pnas.252652399
- Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 2001; 157:103-18; PMID:11139495
- Davis AP, Symington LS. RAD51-dependent break-induced replication in yeast. Mol Cell Biol 2004; 24:2344-51; PMID:14993274; http://dx.doi.org/10.1128/MCB.24.6.2344-2351.2004
- Spencer F, Gerring SL, Connelly C, Hieter P. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics 1990; 124:237-49; PMID:2407610
- Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Gen 1999; 23:81-5; http://dx.doi.org/10.1038/14281
- Zhang C, Roberts TM, Yang J, Desai R, Brown GW. Suppression of genomic instability by SLX5 and SLX8 in Saccharomyces cerevisiae. DNA Repair (Amst) 2006; 5:336-46; PMID:16325482; http://dx.doi.org/10.1016/j.dnarep.2005.10.010
- Myung K, Datta A, Chen C, Kolodner RD. SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nature Gen 2001; 27:113-6; http://dx.doi.org/10.1038/83673
- Rossio V, Galati E, Ferrari M, Pellicioli A, Sutani T, Shirahige K, Lucchini G, Piatti S. The RSC chromatin-remodeling complex influences mitotic exit and adaptation to the spindle assembly checkpoint by controlling the Cdc14 phosphatase. J Cell Biol 2010; 191:981-97; PMID:21098112; http://dx.doi.org/10.1083/jcb.201007025
- Thrower DA, Stemple J, Yeh E, Bloom K. Nuclear oscillations and nuclear filament formation accompany single-strand annealing repair of a dicentric chromosome in Saccharomyces cerevisiae. J Cell Sci 2003; 116:561-9; PMID:12508116; http://dx.doi.org/10.1242/jcs.00251
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998; 14:953-61; PMID:9717241; http://dx.doi.org/10.1002/(SICI)1097-0061(199807)14:10%3c953::AID-YEA293%3e3.0.CO;2-U
- Muzi Falconi M, Piseri A, Ferrari M, Lucchini G, Plevani P, Foiani M. De novo synthesis of budding yeast DNA polymerase alpha and POL1 transcription at the G1/S boundary are not required for entrance into S phase. Proc Natl Acad Sci U S A 1993; 90:10519-23; PMID:8248139; http://dx.doi.org/10.1073/pnas.90.22.10519