
ABSTRACT
Autophagy and senescence are 2 distinct pathways that are importantly involved in acute kidney injury and renal repair. Recent data indicate that the 2 processes might be interrelated. To investigate the potential link between autophagy and senescence in the kidney we isolated primary tubular epithelial cells (PTEC) from wild-type mice and monitored the occurrence of cellular senescence during autophagy activation and inhibition. We found that the process of cell isolation and transfer into culture was associated with a strong basal autophagic activation in PTEC. Specific inhibition of autophagy by silencing autophagy-related 5 (Atg5) counteracted the occurrence of senescence hallmarks under baseline conditions. Reduced senescent features were also observed in Atg5 silenced PTEC after γ-irradiation and during H-Ras induced oncogenic senescence, but the response was less uniform in these stress models. Senescence inhibition was paralleled by better preservation of a mature epithelial phenotype in PTEC. Interestingly, treatment with rapamycin, which acts as an activator of autophagy, also counteracted the occurrence of senescence features in PTEC. While we interpret the anti-senescent effect of rapamycin as an autophagy-independent effect of mTOR-inhibition, the more specific approach of Atg5 silencing indicates that overactivated autophagy can have pro-senescent effects in PTEC. These results highlight the complex interaction between cell culture dependent stress mechanisms, autophagy and senescence.
Introduction
Autophagy is an evolutionarily conserved cellular process which mediates efficient turnover of damaged organelles, proteins and other macromolecules by self-degradation through the hydrolases of lysosomes. Autophagy generally acts as a housekeeping mechanism which is crucial to the maintenance of normal cellular homeostasis. However, when stimulated by cell stress, autophagy may also function as a survival pathway that enables cellular viability under unfavorable conditions. In the kidney, this has been shown in numerous models of acute kidney injury (AKI) such as cisplatin nephrotoxicity and ischemia reperfusion (I/R) injury.Citation1,2 Since autophagy deletion worsened tubular damage and kidney function in AKI it was concluded that autophagy acts as a renoprotective mechanism.Citation2-4
Another mechanism that is triggered by AKI is cellular senescence. Cellular senescence defined as a state of irreversible cell cycle arrest is mediated by cyclin-dependent kinase inhibitors, particularly p16INK4a.Citation5 The expression level of p16INK4a is upregulated in the mammalian kidney during aging and in many forms of kidney disease.Citation6-9 In kidney transplants expression of p16INK4a can serve as a strong predictor of functional allograft outcome.Citation10,11 Based on these observations it has been suggested that cellular senescence is involved in impaired renal repair and in the progression from AKI to chronic kidney disease (CKD).Citation12 In support of this view, we have observed that experimental ablation of p16INK4a-driven senescence leads to a favorable outcome after AKI and to improved graft survival.Citation13 Most recently, Baker and colleagues have shown that clearance of p16INK4a expressing cells extends lifespan and attenuates age dependent glomerulosclerosis.Citation14
Based on the finding that stress-induced autophagy is often followed by the occurrence of senescent cells it was concluded that autophagy might act as a pro-senescent trigger.Citation15-19 Along these lines, Young et al. first proposed autophagy as an effector mechanism for establishment of oncogenic senescence.Citation17 Autophagy was later also found to contribute to senescence of bile duct epithelial cells and progression toward primary biliary cirrhosis.Citation15,16 Perez-Neut et al. showed that autophagy in melanoma cells can function as a pro-survival mechanism by driving senescence induction after pharmacologic potassium channel activation.Citation20 We recently observed that genetic ablation of autophagy in a subset of kidney tubular cells counteracted the development of senescence.Citation21 However, the interrelationship between autophagy and senescence is debated because other studies clearly indicate that autophagy can exert anti-senescent effects.Citation22,23 Down regulation of autophagy in bronchial epithelial cells and in hepatocytes has been shown to accelerate senescence.Citation24,25 More recent data indicate that constitutive autophagy might prevent the activation of pro-senescent pathways in muscle stem cells.Citation23
In the kidney, senescence occurs primarily in tubular epithelial cells,Citation8,14 a highly specialized cell type that is quiescent under normal conditions but has the ability to induce extensive cell cycling in response to acute injury.Citation26 In order to gain better insight regarding the link between autophagy and senescence we isolated cells from the tubular epithelial compartment and tested the impact of autophagy modulation on the occurrence of cellular senescence in vitro.
Results
PTEC have a high baseline autophagic activity
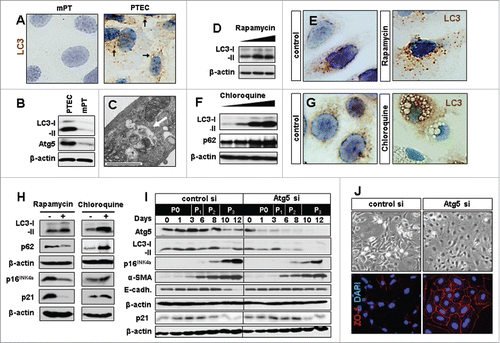
The process of tubular cell isolation and subsequent culturing is associated with stress that drives a senescent phenotype in PTEC.Citation27 In order to define the potential involvement of autophagy in the underlying process we first analyzed the baseline autophagic activity of PTEC. When compared to immortalized proximal tubular cells (mPT), which do not senesce, PTEC displayed massively higher levels of autophagy as shown by LC3-positive punctae, LC3-I/-II conversion and increased expression of autophagy-related 5 (Atg5) (). A further induction of autophagy was observed when PTEC were treated with the mTOR-inhibitor rapamycin (). Treatment with autophagy inhibitor chloroquine also resulted in higher LC3-II levels due to a degradation block as reflected by accumulation of p62 and the abundant occurrence of autophagy vacuoles (). These data indicate a strong baseline autophagic activity in PTEC which can be enhanced or inhibited by standard autophagy modifying compounds.
Figure 1. Experimental modification of baseline autophagy influences senescence marker expression and epithelial phenotype in renal primary tubular epithelial cells (PTEC). In vitro culturing of PTEC induces autophagy. (A) LC3 staining in mouse proximal tubular cells (mPT) cells and PTEC showing LC3 punctae (arrows) which are localized on the autophagosomal membrane during autophagy induction. (B) Representative LC3 immunoblots in PTEC as compared to mPT cells. (C) Electron microscopy image of PTEC with autophagic vacuoles representing baseline autophagy. (D) LC3 immunoblot in PTEC treated with increasing concentrations of rapamycin. (E) Immunocytochemistry for LC3 punctae upon autophagy induction with rapamycin. (F) Representative immunoblots for LC3 and p62 and (G) immunocytochemistry for LC3 aggregates and vacuole formation in PTEC upon autophagy inhibition with increasing concentrations of chloroquine. (H) Immunoblots for LC3, p62, p16INK4a and p21 at day 12 in PTEC treated with rapamycin and chloroquine. (I) Immunoblots for Atg5, LC3, p16INK4a, α-smooth muscle actin (α-SMA), E-cadherin (E-cadh.) and p21 in Atg5 or control siRNA treated PTEC harvested at indicated time points and passages (P0-P3). (J) Representative pictures of day 12 PTEC transfected with control or Atg5 siRNA in bright field and immunohistochemistry for epithelial marker ZO-1. Original magnification x 630 in A, E, and G and x 400 in J. Scale bar 1 µm in C. n = 3.
Impact of experimental autophagy modification on senescence markers and epithelial phenotype under baseline culture conditions
Cellular senescence is characterized by expression of cell cycle inhibitor p16INK4a which is progressively up-regulated in PTEC under baseline culture conditions.Citation27 Autophagy activation by rapamycin was associated with a reduction in p16INK4a and p21 expression whereas chloroquine had a minor, possibly enhancing effect on these proteins (). Although these results might argue for an anti-senescent impact of autophagy, it is important to note that all chemical modifiers of autophagy, including rapamycin and chloroquine, have off-target effects as they affect many basic cellular processes.Citation28 To dissect the potential interrelation between autophagy and senescence in a more specific way we used siRNA based knockdown of Atg5. When Atg5 silenced PTEC were longitudinally followed for 12 days a reduction in LC3-I/-II conversion was observed which was associated with a reduced up-regulation of p16INK4a (). Morphologically, this was paralleled by better preservation of the PTEC epithelial phenotype as reflected by maintenance of ZO-1 and E-cadherin and reduced induction of α-SMA (). No significant differences were found in p21 expression () or senescence-associated β-galactosidase (SA-β-GAL) activity between the groups (Fig. S1A, B, E)
Impact of experimental autophagy modification on senescence markers and epithelial phenotype under pro-senescent stress conditions
We have previously observed that γ-irradiation can be used as an effective strategy to drive PTEC toward accelerated synchronized senescence.Citation27 To examine the impact of γ-irradiation on autophagy, PTEC were irradiated and analyzed by electron microscopy. At day 1 after irradiation large autophagosomal formations were observed (). This was paralleled by enhanced LC3-I/-II conversion at 24 hours which subsequently diminished and was lower at day 10 when compared to control PTEC (). Silencing of Atg5 in γ-irradiated PTEC resulted in a significantly reduced p16INK4a induction (), reduced numbers of γ-H2AX+/Ki67- nuclei and a higher proliferation rate (). While this suggested an attenuated development of senescence, the expression of p21 was elevated and the activity of SA-β-GAL was increased in Atg5 silenced cells (, Fig. S1C, D, F). In parallel, there was a reduced upregulation of α-SMA and better maintenance of E-cadherin expression reflecting a preserved epithelial phenotype (). Interestingly, treatment of γ-irradiated PTEC with rapamycin and the associated activation of autophagy also attenuated the upregulation of p16INK4a and p21 (). Inhibition of autophagy with chloroquine revealed a marginal increase in p16INK4a and p21 expression () but this effect has to be interpreted with caution as the combination of γ-irradiation with chloroquine treatment strongly affected PTEC viability.
Figure 2. Experimental modification of autophagy influences senescence marker expression and epithelial phenotype under pro-senescent stress conditions in renal primary tubular epithelial cells (PTEC). PTEC underwent established models of senescence induction using γ-irradiation (10 Gy) or retrovirus-mediated overexpression of constitutively active mutant H-Ras. In parallel, autophagy was suppressed by siRNA mediated knock down of Atg5. (A) Representative electron microscopy images from PTEC showing (a) presence of massive autophagy related structures (defined as early double membraned structures surrounding cellular content as well as later stages, where the inner membrane has already become indiscernible) at day 1 (b) a decline in autophagy at day 10 after γ-irradiation. (B) Immunoblots for Atg5, LC3, p16INK4, α-smooth muscle actin (α-SMA), E-cadherin (E-cadh.) and p21 in γ-irradiated PTEC treated with Atg5 or control siRNA. (C, D) Quantification of γ-H2AX+/Ki67− cells and BrdU incorporation in γ-irradiated PTEC treated with Atg5 or control siRNA. (E, F) Representative immunoblots for LC3, p62, p16INK4a and p21 in γ-irradiated PTEC treated with rapamycin or chloroquine. (G) Representative electron microscopy images from PTEC showing presence of large autophagy related structures at day 6 after H-Ras transduction. (H) Immunoblots for H-Ras, p16INK4a and p21 expression in Atg5 siRNA treated H-Ras transduced PTEC. (I, J) Quantification of γ-H2AX+/Ki67− cells and BrdU incorporation in H-Ras transduced PTEC after Atg5 or control siRNA treatment. (K) Representative immunoblots for H-Ras, p16INK4a and p21 upon rapamycin treatment in H-Ras transduced PTEC. Scale bar 1 µm in A and G. Data is presented as mean ± SEM. n = 7. *P < 0.05; **P < 0.005; ***P < 0.001.
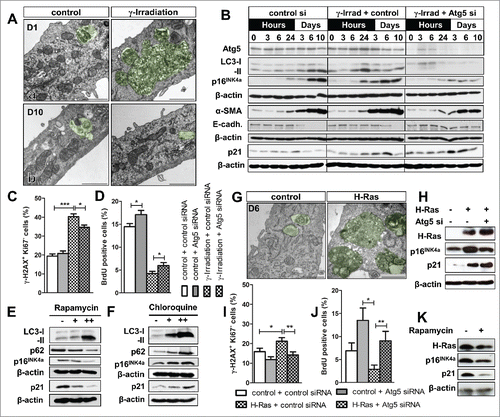
In order to extend our findings to a different model of senescence induction we investigated the impact of autophagy in PTEC undergoing oncogenic H-Ras-dependent senescence. As previously described in other cell types H-Ras transduction was associated with enhanced induction of autophagosomes and in parallel resulted in an up-regulation of p16INK4a (). This effect on p16INK4a expression was reduced when autophagy was inhibited via Atg5 silencing (). In line with an attenuated induction of pro-senescent mechanisms Atg5 silencing was also associated with a reduced number of γ-H2AX+/Ki67- nuclei and better proliferation capacity (). Expression of p21, however, was not reduced in Atg5 silenced PTEC (). Similar to our results under baseline conditions and after γ-irradiation, treatment with rapamycin attenuated the H-Ras induced upregulation of p16INK4a and p21 (). The combination of H-Ras transduction and chloroquine treatment could not be studied in PTEC due to excessive cell death.
Discussion
In this study we found that PTEC had a strong basal autophagic activity. Genetic inhibition of autophagy decelerated the occurrence of key senescence markers. These findings are consistent with other reports showing that autophagy can trigger pro-senescent events when activated in response to cellular stress.Citation15,19,21,29 Constitutive baseline autophagy, on the other hand, seems to act usually in a senescence preventing manner playing a crucial role in healthy aging and life span extension.Citation23,30
In our previous work we have shown a rapid development of senescence in PTEC not only after γ-irradiation but also under baseline conditions.Citation27 As described for other primary cell types PTEC undergo an inevitable ‘culture shock’ which refers to the process of cell isolation, the transfer into culture and the conditions of an artificial environment.Citation31 Our current data in PTEC indicate that senescence occurring under this culture shock is preceded by a strong induction of autophagy. Although inhibition of autophagy led to a reduction in the occurrence of key senescence features, SA-β-GAL activity was not affected under baseline conditions and increased when Atg5-silencing was combined with γ-irradiation. It is important to note, that currently there is no singular biomarker that can effectively differentiate between normal and senescent cells. Instead, identification of the senescent state relies on detection of a combination of hallmark features. SA-β-GAL is a frequently used biomarker but it is known for its low specificity and enhanced activity during quiescence and in response to various forms of stress.Citation32 In our experiments we used cell-culture associated stress as the baseline pro-senescent factor. The additional senescence triggers, i.e. γ-irradiation and oncogenic H-Ras expression, which were superimposed in some experiments, are potent activators of the DNA damage response (DDR) pathway. The heterogeneity between SA-β-GAL results might thus reflect different involvement of DDR activation which has been identified as an important factor to determine whether autophagy counteracts or favors senescence.Citation33
Interestingly, we found that not only genetic silencing of autophagy resulted in bypassing senescence marker expression but also the treatment with rapamycin which is commonly used as an activator of autophagy. Given the superior specificity of Atg5 siRNA interference as an autophagy modulating approach our data strongly suggest that the senescence postponing effect of rapamycin was due to autophagy-independent actions of mTOR-inhibition. It has been shown that rapamycin leads to bypassing senescence in primary rodent cells through activated transcription of pluripotent genes, e.g. oct-4, sox-2 and nanog, which is unrelated to autophagy.Citation34 The lack of specificity of rapamycin and other chemical agents, such as chloroquine, has been acknowledged and might be the most important weakness in many autophagy reports.Citation35,36 Chloroquine is an anti-malaria drug, which blocks lysosome function and has therefore a major impact on cell growth and survival.Citation37 We observed that treatment with chloroquine was associated with considerably higher cell damage and cell death in PTEC which precluded studies in combination with γ-irradiation and H-Ras associated stress.
During normal aging accumulation of senescent cells is a slow process. However, p16INK4a reporter mice have revealed that the occurrence of senescent cells can be strongly accelerated by exposure to a variety of environmental toxicants.Citation38 Rapid occurrence of senescence has also been found in patients who had undergone irradiation or chemotherapy.Citation5,39 In the kidney, we and others have previously observed that acute stress, such as experimental AKI by I/R, can trigger a forced development of cellular senescence.Citation13,40,41 Tubular epithelial cells are the Achilles heel in most forms of AKI because they are the most sensitive cell type to hypoxic or toxic damage. Hagemann and colleagues suggested that tubular cell isolation in itself which is associated with a high level of oxidative, ischemic and mechanical stress could be a useful model of AKI.Citation42 Similar to our own experience they described that the outgrowth of PTEC from freshly isolated tubular fragments is characterized by a period of strong proliferation and subsequent cellular differentiation toward a mature epithelial phenotype. However, when isolated PTEC are passaged and followed for longer periods their epithelial phenotype is progressively lost and the cells start to express mesenchymal markers, e.g., α-SMA. Importantly, we observed that this in vitro epithelial-mesenchymal transition (EMT) was postponed in PTEC when Atg5 was silenced. We cannot conclude from our experiments whether this phenotypical change is senescence dependent or, alternatively, a direct effect of autophagy inhibition. An EMT promoting effect of autophagy was shown in different cancer cells.Citation43,44 On the other hand, an anti-EMT role of autophagy sustaining the epithelial phenotype has been described in hepatocytes and in different organs during embryogenesis.Citation45,46 We found reduced signs of epithelial dedifferentiation in post-ischemic kidneys when Atg5 was conditionally ablated from proximal tubular S3 segments.Citation21 The kidneys also developed less post-injurious senescence arguing for a potential link between epithelial phenotypical maintenance, autophagy and senescence.
The present study investigated the impact of autophagy on stress induced senescence in vitro. This type of cell cycle arrest which can be observed in mouse and in human primary cells is independent of telomere length.Citation47 Telomere dependent senescence cannot be adequately studied in murine cells, which, in contrast to most human cells, have much longer telomeres and often express telomerase.Citation47 However, a link between telomere attrition, senescence and autophagy has recently been demonstrated in telomerase deficient mice that showed delayed recovery after kidney I/R.Citation48 It was concluded that renal tubular senescence and subsequent autophagic impairment may synergistically contribute to delayed kidney repair. However, since other data failed to show an interaction between telomere dysfunction-driven senescence and autophagyCitation49 future studies in human PTEC might be instrumental to elucidate the impact of autophagy on telomere-dependent senescence.
In conclusion, our data suggest a functional link between stress-induced autophagy and stress-induced senescence. Culture conditions in PTEC were associated with a high basal rate of autophagy. Specific inhibition of this strong autophagic activity by Atg5 silencing counteracted the occurrence of several markers of PTEC senescence and of epithelial phenotypical loss under baseline conditions and after γ-irradiation and H-Ras overexpression. These results argue for a complex interaction between the processes of autophagy and senescence, in which an overactivation of the autophagic pathway may have disruptive effects for cellular maintenance. The underlying mechanisms that orchestrate the outcome of this interaction are context dependent and may vary with the individual trigger of these 2 important homeostatic cell responses.
Materials and methods
Cell culture
Primary tubular epithelial cells (PTEC) from C57BL/6J mice were isolated as previously described.Citation50 In brief, mice were anesthetized with isoflurane and kidneys were harvested after cardial perfusion with Hanks 199 medium (22340-020, Gibco) with 0,125% Collagenase Type I (138201 GM, Affymetrix). Kidneys were digested at 37°C in a bubble-agitated collagenase solution for 40 minutes and tubules were then separated by size in a 40 µM cell strainer (352340, BD ) and cultured in REGM-II medium (C-26030, Promocell GmbH). Coordinated senescence was induced using 10 Grays of γ-irradiation as previously described.Citation27 Additionally, senescent PTEC were generated by retrovirus-mediated overexpression of constitutively active mutant form of H-Ras oncogene (H-RasV12) using MSCV H-Ras-V12-IRES-GFP (18780, Addgene) and control plasmid MSCV-IRES-GFP (20672, Addgene). Post-irradiation or after H-Ras overexpression, PTECs were cultured for 10 days. In addition, PTEC were allowed to undergo replicative senescence in culture by passaging. For siRNA mediated knock down experiments, Atg5 siRNA (SASI_Mm01_00089196, Sigma-Aldrich) or universal negative control siRNA (SIC001, Sigma-Aldrich) was transfected using Lipofectamine RNAiMAx reagent (13378, Invitrogen) according to manufacturer's protocol. Efficiency of knockdown was confirmed by immunoblotting. For proliferation assays, cells were exposed to 20 μM 5-bromodeoxyuridine (11296736001, Roche) for 4 hrs. A cell line of mouse proximal tubular cells (mPT) was kindly provided by Dr. Lloyd Cantley, Yale School of Medicine. All experimental methods were in agreement with the institutional guidelines for animal research and were approved by the state authorities.
Immunocytochemistry and Senescence-associated-β-galactosidase (SA-β-GAL) staining
Cells were fixed in 4 % buffered PFA and permeabilized by 0.1 % Triton in phosphate-buffered saline (PBS), blocked with 5 % milk and fluorescently stained with anti-BrdU (B8434, Sigma-Aldrich), anti-ZO-1 (61-7300, Invitrogen), anti-Ki67 (SP6, Thermo Fisher Scientific), anti-γ-H2AX (JBW301, EMD Millipore) and LC3 (5F10, Nanotools). The percentage of positive cells was quantified by counting average positive cells in 20 random, non-overlapping HPFs (x400). For assessing SA-β-GAL activity, cells were fixed in a fixation solution comprising of 2% formaldehyde and 0.2% glutaraldehyde in PBS at room temperature for 10 min. Following rinsing with PBS, the cells were incubated over night at 37°C with the X-GAL staining solution pH 6.0 (40 mM citric acid/Na phosphate buffer, 5 mM K4[Fe(CN)6] 3H2O, 5 mM K3[Fe(CN)6], 150 mM sodium chloride, 2 mM magnesium chloride and 1 mg/ml X-gal in distilled water). The cells were viewed by bright field microscopy and percentage of positive cells was quantified by counting average number of positive cells in 8 random HPFs (X200).
Ultrastructural analysis
PTEC were fixed in a mixture of 150 mM HEPES, pH 7.35, containing 1.5% formaldehyde and 1.5% glutaraldehyde and immobilized in agarose. Agarose blocks (2 mm3 blocks) were postfixed in 1% OsO4 with 1.5% potassium ferrocyanide, respectively, and 4% aqueous uranyl acetate. Samples were dehydrated in acetone and embedded in Epon (Agar 100 resin, Agar Scientific). Ultrathin sections (50 nm) were post stained with uranyl acetate and lead citrate and observed in a Morgagni transmission electron microscope (FEI, Eindhoven, Netherlands). Images were procured with a side mounted Veleta CCD camera.
Western blotting
Immunoblot was performed as previously described.Citation50 Protein lysates were electrophoresed on SDS-PAGE and were transferred to PVDF membrane. The membranes were blocked with 5% milk in Tris-buffered saline with Tween-20, and incubated overnight at 4˚C with primary antibodies: anti-LC3 B-II (L8918, Sigma-Aldrich or 2775, Cell Signalling), anti-p62 (p0067, Sigma-Aldrich), anti-Ras (3965, Cell Signalling), anti-p16INK4a (SC-1207, Santa Cruz), anti-p21 (556430, BD Biosciences), anti-E-cadherin (24E10, Cell Signalling), α-smooth muscle actin (A5228, Sigma-Aldrich) and anti-β-actin (ab82618, Abcam). Post incubation, membranes were incubated with appropriate HRP-conjugated secondary antibodies (7074s and 7076s, Cell Signalling) and were visualized by Immobilon Western Chemiluminescent HRP Substrate (RPN2106, EMD Millipore).
Statistical analysis
Results are expressed as means ± SEM. Statistical significance was determined by unpaired t test (GraphPad Software, San Diego, CA). P < 0.05 was considered to be statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary File
Download TIFF Image (241.3 KB)Acknowledgments
Parts of this work have been presented at the ASN Renal Week 2014, Philadelphia and at the Experimental Biology Conference 2015, Boston.
Funding
This study was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich SFB 738 to AM and RS and Dr. Werner Jackstädt-Stiftung to RS).
ORCID
Christoph Wrede http://orcid.org/0000-0001-7461-6965
Related Research Data
References
- Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 2012; 180:517-25; PMID:22265049; http://dx.doi.org/10.1016/j.ajpath.2011.11.001
- Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011; 22:902-13; PMID:21493778; http://dx.doi.org/10.1681/ASN.2010070705
- Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int 2016; 89:779-91; PMID:26924060; http://dx.doi.org/10.1016/j.kint.2015.11.021
- Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 2012; 82:1271-83; PMID:22854643; http://dx.doi.org/10.1038/ki.2012.261
- Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 2015; 21:1424-35; PMID:26646499; http://dx.doi.org/10.1038/nm.4000
- Melk A, Schmidt BM, Vongwiwatana A, Rayner DC, Halloran PF. Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am J Transplant 2005; 5:1375-82; PMID:15888044; http://dx.doi.org/10.1111/j.1600-6143.2005.00846.x
- Sis B, Tasanarong A, Khoshjou F, Dadras F, Solez K, Halloran PF. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int 2007; 71:218-26; PMID:17183247; http://dx.doi.org/10.1038/sj.ki.5002039
- Melk A, Schmidt BM, Takeuchi O, Sawitzki B, Rayner DC, Halloran PF. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int 2004; 65:510-20; PMID:14717921; http://dx.doi.org/10.1111/j.1523-1755.2004.00438.x
- Melk A, Kittikowit W, Sandhu I, Halloran KM, Grimm P, Schmidt BM, Halloran PF. Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int 2003; 63:2134-43; PMID:12753300; http://dx.doi.org/10.1046/j.1523-1755.2003.00032.x
- Gingell-Littlejohn M, McGuinness D, McGlynn LM, Kingsmore D, Stevenson KS, Koppelstaetter C, Clancy MJ, Shiels PG. Pre-transplant CDKN2A expression in kidney biopsies predicts renal function and is a future component of donor scoring criteria. PLoS One 2013; 8:e68133; PMID:23861858; http://dx.doi.org/10.1371/journal.pone.0068133
- Koppelstaetter C, Schratzberger G, Perco P, Hofer J, Mark W, Ollinger R, Oberbauer R, Schwarz C, Mitterbauer C, Kainz A, et al. Markers of cellular senescence in zero hour biopsies predict outcome in renal transplantation. Aging Cell 2008; 7:491-7; PMID:18462273; http://dx.doi.org/10.1111/j.1474-9726.2008.00398.x
- Schmitt R, Melk A. New insights on molecular mechanisms of renal aging. Am J Transplant 2012; 12:2892-900; PMID:22882799; http://dx.doi.org/10.1111/j.1600-6143.2012.04214.x
- Braun H, Schmidt BM, Raiss M, Baisantry A, Mircea-Constantin D, Wang S, Gross ML, Serrano M, Schmitt R, Melk A. Cellular senescence limits regenerative capacity and allograft survival. J Am Soc Nephrol 2012; 23:1467-73; PMID:22797186; http://dx.doi.org/10.1681/ASN.2011100967
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016; 530:184-9; PMID:26840489; http://dx.doi.org/10.1038/nature16932
- Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest 2010; 90:835-43; PMID:20212459; http://dx.doi.org/10.1038/labinvest.2010.56
- Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy may precede cellular senescence of bile ductular cells in ductular reaction in primary biliary cirrhosis. Dig Dis Sci 2012; 57:660-6; PMID:21989821; http://dx.doi.org/10.1007/s10620-011-1929-y
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM. Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23:798-803; PMID:19279323; http://dx.doi.org/10.1101/gad.519709
- Liu H, He Z, Simon HU. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 2014; 10:372-3; PMID:24300435; http://dx.doi.org/10.4161/auto.27163
- Lenain C, Gusyatiner O, Douma S, van den Broek B, Peeper DS. Autophagy-mediated degradation of nuclear envelope proteins during oncogene-induced senescence. Carcinogenesis 2015; 36:1263-74; PMID:26354777; http://dx.doi.org/10.1093/carcin/bgv124
- Perez-Neut M, Haar L, Rao V, Santha S, Lansu K, Rana B, Jones WK, Gentile S. Activation of hERG3 channel stimulates autophagy and promotes cellular senescence in melanoma. Oncotarget 2016; 7:21991-2004; PMID:26942884
- Baisantry A, Bhayana S, Rong S, Ermeling E, Wrede C, Hegermann J, Pennekamp P, Sorensen-Zender I, Haller H, Melk A, et al. Autophagy Induces Prosenescent Changes in Proximal Tubular S3 Segments. J Am Soc Nephrol 2015; PMID:AMBIGUOUS
- Grootaert MO, da Costa Martins PA, Bitsch N, Pintelon I, De Meyer GR, Martinet W, Schrijvers DM. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015; 11:2014-32; PMID:26391655; http://dx.doi.org/10.1080/15548627.2015.1096485
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. Autophagy maintains stemness by preventing senescence. Nature 2016; 529:37-42; PMID:26738589; http://dx.doi.org/10.1038/nature16187
- Toshima T, Shirabe K, Fukuhara T, Ikegami T, Yoshizumi T, Soejima Y, Ikeda T, Okano S, Maehara Y. Suppression of autophagy during liver regeneration impairs energy charge and hepatocyte senescence in mice. Hepatology 2014; 60:290-300; PMID:24668739; http://dx.doi.org/10.1002/hep.27140
- Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, Minagawa S, Yumino Y, Ishikawa T, Numata T, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012; 1:630-41; PMID:22934255; http://dx.doi.org/10.4161/onci.20297
- Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int 2009; 76:604-13; PMID:19536080; http://dx.doi.org/10.1038/ki.2009.224
- Berkenkamp B, Susnik N, Baisantry A, Kuznetsova I, Jacobi C, Sorensen-Zender I, Broecker V, Haller H, Melk A, Schmitt R. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS One 2014; 9:e88071; PMID:24505380; http://dx.doi.org/10.1371/journal.pone.0088071
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/10.1038/nrd3802
- Chang TC, Hsu MF, Wu KK. High glucose induces bone marrow-derived mesenchymal stem cell senescence by upregulating autophagy. PLoS One 2015; 10:e0126537; PMID:25961745; http://dx.doi.org/10.1371/journal.pone.0126537
- Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest 2015; 125:85-93; PMID:25654554; http://dx.doi.org/10.1172/JCI73946
- Sherr CJ, DePinho RA. Cellular senescence: mitotic clock or culture shock? Cell 2000; 102:407-10; PMID:10966103; http://dx.doi.org/10.1016/S0092-8674(00)00046-5
- Matjusaitis M, Chin G, Sarnoski EA, Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing Res Rev 2016; 29:1-12; PMID:27212009; http://dx.doi.org/10.1016/j.arr.2016.05.003
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015; 349:aaa5612; PMID:26404840; http://dx.doi.org/10.1126/science.aaa5612
- Pospelova TV, Bykova TV, Zubova SG, Katolikova NV, Yartzeva NM, Pospelov VA. Rapamycin induces pluripotent genes associated with avoidance of replicative senescence. Cell Cycle 2013; 12:3841-51; PMID:24296616; http://dx.doi.org/10.4161/cc.27396
- Gewirtz DA. Autophagy and senescence: a partnership in search of definition. Autophagy 2013; 9:808-12; PMID:23422284; http://dx.doi.org/10.4161/auto.23922
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Farkas T, Daugaard M, Jaattela M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J Biol Chem 2011; 286:38904-12; PMID:21930714; http://dx.doi.org/10.1074/jbc.M111.269134
- Sorrentino JA, Krishnamurthy J, Tilley S, Alb JG, Jr., Burd CE, Sharpless NE. p16INK4a reporter mice reveal age-promoting effects of environmental toxicants. J Clin Invest 2014; 124:169-73; PMID:24334456; http://dx.doi.org/10.1172/JCI70960
- Marcoux S, Le ON, Langlois-Pelletier C, Laverdiere C, Hatami A, Robaey P, Beausejour CM. Expression of the senescence marker p16INK4a in skin biopsies of acute lymphoblastic leukemia survivors: a pilot study. Radiat Oncol 2013; 8:252; PMID:24171943; http://dx.doi.org/10.1186/1748-717X-8-252
- Chkhotua AB, Abendroth D, Froeba G, Schelzig H. Up-regulation of cell cycle regulatory genes after renal ischemia/reperfusion: differential expression of p16(INK4a), p21(WAF1/CIP1) and p27(Kip1) cyclin-dependent kinase inhibitor genes depending on reperfusion time. Transpl Int 2006; 19:72-7; PMID:16359379; http://dx.doi.org/10.1111/j.1432-2277.2005.00227.x
- Westhoff JH, Schildhorn C, Jacobi C, Homme M, Hartner A, Braun H, Kryzer C, Wang C, von Zglinicki T, Kranzlin B, et al. Telomere shortening reduces regenerative capacity after acute kidney injury. J Am Soc Nephrol 2010; 21:327-36; PMID:19959722; http://dx.doi.org/10.1681/ASN.2009010072
- Hagemann JH, Thomasova D, Mulay SR, Anders HJ. Nrf2 signalling promotes ex vivo tubular epithelial cell survival and regeneration via murine double minute (MDM)-2. Nephrol Dial Transplant 2013; 28:2028-37; PMID:23476038; http://dx.doi.org/10.1093/ndt/gft037
- Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, Song Z, Zheng Q, Xiong J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013; 34:1343-51; PMID:23430956; http://dx.doi.org/10.1093/carcin/bgt063
- Zou M, Zhu W, Wang L, Shi L, Gao R, Ou Y, Chen X, Wang Z, Jiang A, Liu K, et al. AEG-1/MTDH-activated autophagy enhances human malignant glioma susceptibility to TGF-beta1-triggered epithelial-mesenchymal transition. Oncotarget 2016; 7:13122-38; PMID:26909607
- Lu WH, Wang G, Li Y, Li S, Song XY, Wang XY, Chuai M, Lee KK, Cao L, Yang X. Autophagy functions on EMT in gastrulation of avian embryo. Cell Cycle 2014; 13:2752-64; PMID:25486362; http://dx.doi.org/10.4161/15384101.2015.945850
- Grassi G, Di Caprio G, Santangelo L, Fimia GM, Cozzolino AM, Komatsu M, Ippolito G, Tripodi M, Alonzi T. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis 2015; 6:e1880; PMID:26355343; http://dx.doi.org/10.1038/cddis.2015.249
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev 2010; 24:2463-79; PMID:21078816; http://dx.doi.org/10.1101/gad.1971610
- Cheng H, Fan X, Lawson WE, Paueksakon P, Harris RC. Telomerase deficiency delays renal recovery in mice after ischemia-reperfusion injury by impairing autophagy. Kidney Int 2015; 88:85-94; PMID:25760322; http://dx.doi.org/10.1038/ki.2015.69
- Mar FA, Debnath J, Stohr BA. Autophagy-independent senescence and genome instability driven by targeted telomere dysfunction. Autophagy 2015; 11:527-37; PMID:25751002; http://dx.doi.org/10.1080/15548627.2015.1017189
- Schmitt R, Marlier A, Cantley LG. Zag expression during aging suppresses proliferation after kidney injury. J Am Soc Nephrol 2008; 19:2375-83; PMID:18815245; http://dx.doi.org/10.1681/ASN.2008010035