
ABSTRACT
Directed cell conversion (or transdifferentiation) of one somatic cell-type to another can be achieved by ectopic expression of a set of transcription factors. Since the experimental identification of transcription factors for transdifferentiation is extremely time-consuming and expensive, there are still relatively few transdifferentiations achieved in comparison to the number of human cell-types. However, the growing volume of transcriptional data available and the recent introduction of data-driven algorithmic approaches that predict factors for transdifferentiation holds great promise for accelerating this field. Here we review those computational methods whose in-silico predictions have been experimentally validated, highlighting differences and similarities. Our analysis reveals that the factors predicted by each method tend to be different due to varying source cells used, gene expression quantification and algorithmic steps. We show these differences have an impact on the regulatory influences downstream, with some methods favoring transcription factors regulating developmental progression and others favoring factors regulating mature cell processes. These computational approaches offer a starting point to predict and test novel factors for transdifferentiation. We argue that collecting high-quality gene expression data from single-cells or pure cell-populations across a broader set of cell-types would be necessary to improve the quality and consistency of the in-silico predictions.
Introduction
Our understanding of cell phenotype and its state has been transformed over the last decade as it has become increasingly clear that cell-type is not necessarily fixed. The process of experimentally inducing changes in cellular identity by ectopic factors such as transcription factors (TFs), drugs, growth factors, chemicals, etc. is known as direct cell conversion. Furthermore, transdifferentiation is defined as the process of converting the cells from one somatic lineage to another, and in this review the terms ‘cell conversion’ and ‘transdifferentiation’ will be used interchangeably. These phenomena were initially observed in a cell transition from fibroblasts to myoblasts caused by the overexpression of transcription factor MYOD in fibroblasts.Citation1 More recently, Shinya Yamanaka famously demonstrated that a small set of TFs, namely OCT4, KLF4, SOX2 and MYC can be used to transform human fibroblasts to induced pluripotent stem cell (iPS).Citation2 Subsequently, other groups demonstrated that it was possible to convert fibroblasts to hepatocytes,Citation3,4,5 to cardiomyocyteCitation6,7,8 and to several other cell-types (for reviews see refs.Citation9,10,11). Despite these breakthroughs, the rate at which new sets of TFs for transdifferentiation has been discovered has been slow, likely owing to the difficulty of identifying these factors by the “trial and error” approach taken by experimental groups. To overcome the limitation of these strategies, a number of computational techniques have recently emerged with the aim of speeding up the discovery of TFs sets, for instance Heinaniemi M et al.,Citation12 Lang AH et al.,Citation13 Crespo I et al.,Citation14 Davis FP et al.,Citation15 D'Alessio AC et al.,Citation16 CellNetCitation17 and MogrifyCitation18 (reviewed by Bian Q and Cahan P19). Here, we will focus on the 3 computational methods whose TF predictions have been experimentally validated via human cell conversions; CellNetCitation17 which uses microarray gene expression data and correlation-based network score, D'Alessio et al.Citation16 whose method uses microarray gene expression data and an information theoretic approach (Jensen-Shannon Divergence; from herein we will refer to this method as JSD) and MogrifyCitation18 which uses cap analysis of gene expression (CAGE) data and a network based score. Each of these computational methods uses information on cell-type specific gene expression, and in some cases gene-regulatory network information, as their input to calculate a score for each TF and output a set (or a ranked list) of TFs for a given cell conversion. With so few known cell conversions and the fact that the same conversion can be achieved with slightly different sets of TFs, it is difficult to assess which of these approaches makes the most reliable predictions without undertaking an extensive experimental validation program. However, here we compared and contrasted the predictions made by each of the approaches across many cell conversions and in doing so we highlight the differences and similarities between the methods. Additionally, we also show that the predictions are sensitive to both the input data and expression profiling technique. Further to this by investigating the molecular networks and functions that the predicted set of TFs regulate, we also investigate in detail the TF predictions from each method for 2 well-characterized cell conversions: fibroblasts to cardiomyocytes-like cells and fibroblasts to hepatocytes-like cells.
Computational methods for the prediction of reprogramming factors
The available methods vary in a number of ways, for instance the input data used, the algorithm and the breadth of predictions available. A schematic summary of each method is reported in . In order to predict TFs for transdifferentiation all methods broadly follow the same computational stages: (1) They collate information about the gene expression profiles of multiple cell-types, (2) For each cell conversion they create a differential gene expression profile between the target cell-type and a background cell-types, (3) Based on the gene expression they assess the transcriptional and regulatory influence of each TF on the target cell-type, (4) They rank TFs based on their relative regulatory influence and (5) Provide an output including the prioritized set of TFs for a given transdifferentiation.
Figure 1. (A) Summary of methods. Each row represents the method for predicting TFs in transdifferentiation which are CellNet, JSD and Mogrify. Each column represents the computational stages involved in the TFs set prediction which are input requirement, generation of differential expression profile, identifying the influence of each TF in cell conversion, criteria to prioritize the TFs and finally the predictions. CellNet classifies the source cell-type based on the user experimental microarray data and provides a ranked list of TFs for conversion. It does this by creating GRN (gene regulatory network) from GEO (gene expression omnibus) data using CLR (context likelihood of relatedness) algorithm and the target cell specific sub-network is obtained by GSA (gene set analysis). The TF influence score is calculated by the differential expression of the TF and the number of genes it regulates. Finally, CellNet outputs classification of the source cell, a list of TFs ranked by their importance in conversion and the target cell-type GRN. As input JSD method requires only the target cell-type detail. The differential expression profile is detected by comparing the target cell profile with the background data set (selected by low Pearson correlation between the expression profiles) using microarray data from GEO. Jensen-Shannon divergence (JSD) is used to measure the deviance of the observed (TF expression profile observed in data from GEO) from the ideal (TF is highly expressed only in target cell compared to the background cells) distribution. Finally, it provides a ranked list of TFs based on the JSD score and specifies top 10 as core candidate TFs. As input Mogrify method requires both source and target cell-type details. The differential expression profile between target cell-type and background dataset (all cell-types excluding the target cell-type) is obtained using the CAGE data (cap analysis of gene expression) from FANTOM5 consortium. A gene interaction network is formed with data from STRING database and MARA. Then the TFs are ranked based on differential expression of the TF, differential expression of the regulated genes and connectivity of the TF in the network. Finally, TFs expressed in the source cell are removed and a non-redundant TFs set is provided. B) Comparison of TFs used for prediction by each method and TFClass which comprises of hierarchical classification of TFs. C) Comparison of target cell-types available for the conversion from fibroblast by each method.
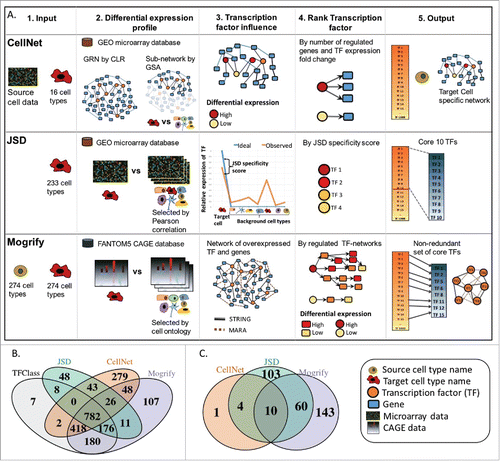
Out of the 3 algorithms, JSD is the only method that does not take into account the source cell-type in its calculations. As a result, the predicted TFs by JSD for transdifferentiation to a particular target cell-type will always be the same regardless of the source cell-type. Conversely, both CellNet and Mogrify take into account the starting cell-type; Mogrify removes TFs if they are already expressed in the source cell-type and CellNet weights its predictions based on the change in expression from the source to the target cell-type.
For transdifferentiation, each of the techniques provide predictions for a different number of cell/tissue-types; CellNet can provide predictions for 16 cell-types, and the software is publically available to run predictions for new cell-types based on user's data. Neither JSD's or Mogrify's software is publically available, but they provide TFs predictions for transdifferentiations for 233 and 274 cell/tissue-types, respectively.
All approaches considered here use gene expression data as the basis to make their predictions, and the data quality will impact the accuracy of the predictions. Both CellNet and JSD use microarray data from the gene expression omnibus (GEO) databaseCitation20, whereas Mogrify uses CAGE data from the FANTOM5 (functional annotation of mammalian).Citation21 Since CAGE is a sequencing-based technology it provides a digital quantification of expressed genes; however, microarrays have issues with reliably distinguishing expression levels, particularly in low abundance genesCitation22 such as TFs.
An important step common to all methods is the calculation of a differential gene expression statistics from gene expression profiles for each of the available cell-types. In order to do this, each method defines a background set of gene expression profiles against which the target gene expression profile is compared. CellNet creates this background dataset by using all the cell-types excluding the target cell-type. JSD creates a static background data set comprising representative cell-types whose Pearson correlations are less than 0.9 and this background is the same irrespective of the target cell-type. Mogrify creates a background dataset based on the FANTOM5 cell-type ontology tree, for instance the background for a target tissue-type comprises of only other tissue-types and for a target cell-type it is made of other cell-types excluding the target cell/tissue-type.
Once the background is selected each approach has a different algorithm for predicting the sets of TFs that can induce a cell conversion. CellNet finds subnetworks using the InfoMap community-detection algorithmCitation23 and then uses GSACitation24 to identify target cell-type specific subnetworks of genes (these genes are highly expressed when compared to all other cell-types). Following this, a TF influence score is computed by a weighted sum of differential expression of the TF and the number of genes it regulates. JSD calculates the Jensen-Shannon Divergence, a deviance measure of observed TF expression with respect to an idealised TF expression across all cell-types. The idealised TF expression is defined as a case where the TF is highly expressed in a target cell and not expressed in any of the background cells. The resulting divergence score is used to rank TFs, with those with the lowest divergence (most specific) being the top predicted TFs. Mogrify creates a score for every gene in each sample by combining the log fold change and adjusted p-value when compared to the background using DESeq.Citation25 Subsequently, to compute a score for each TF a weighted sum of its local network neighborhood defined gene/protein interaction from STRINGCitation26 database and MARA.Citation27 The sum is performed with a weight placed on the number of genes connected to the TF to penalise highly connected TFs and on the distance of regulated gene from the TF of interest, to account for the fact that direct regulation is more effective than indirect. Furthermore, Mogrify has a step that is not present in either of the techniques whereby it calculates which TFs are redundant to each other, and in doing so it calculates an optimal set of transcription factors for each conversion. In contrast, JSD selects the top 10 TFs as candidates and CellNet ranks all TFs for each conversion leaving the choice of how many TFs to choose to the user.
For all 3 methods, the authors of the study provide experimental validation to support the predicted TFs. CellNet improved upon existing transdifferentiations from b-cell to macrophage and also from fibroblast to hepatocyte, JSD performed a transdifferentiation of fibroblast to retinal pigment epithelial cell, and Mogrify carried out 2 cell conversions between fibroblast and keratinocyte as well as between keratinocyte and microvascular endothelial cell. Here we aim to identify the characteristics of TFs predicted by each method, suggest possible biological implications of the TFs based on the genes that they regulate and highlight significant differences between the methods and underlying data that can have an effect on the predictions.
Comparison of methods
Comparing the definition of transcription factor between methods
The breadth of TFs-predictions for each technique can be measured in 2 ways. Firstly, the numbers of cell-types for which predictions are available; which are 16, 233 and 274 cell/tissue-types for CellNet, JSD and Mogrify, respectively. Secondly, the set of potential TFs that can be predicted, which is based on the definition of transcription factor used by each method. CellNet lists a possible 1,599 TFs by selecting genes that have a gene ontology annotation as ‘nucleic acid binding’ and ‘transcriptional regulation’. JSD lists 1,095 TFs and Mogrify lists 1,749 however, neither of these methods provides the selection criteria used to classify TFs. In order to investigate the set of TFs utilized by each method, we compared the TF lists used with a list of genes defined by TFClassCitation28 as transcription factors. TFClass provides a hierarchical classification for 1,573 human TFs based on DNA-binding domains. There are 809 common TFs between the 3 methods, out of which 782 are also defined as TF in TFClass (). Furthermore, 24.7%, 11.6% and 11% of the genes defined as TF by CellNet, JSD and Mogrify respectively, do not appear in TFClass (). Moreover, there are 278 unique TFs that can only be predicted by Mogrify, 281 for CellNet and 56 for JSD. This highlights how differences in the basic definition of TF by each of the techniques can have a large effect on the possible range of TFs being predicted.
Comparing TF-predictions across methods using a tree of cell-types
In order to compare the shared and unique TFs predicted for common conversions by each method, the same/similar cell-types from each technique were mapped onto a cell-type ontology tree. This tree was derived from the UBERONCitation29 anatomical ontology network which describes the relationship between body parts, organs, tissues and cells. The complete cell-type ontology tree of 320 cell-types representing all 3 methods is presented in Fig S1, and a subset of this tree is shown in . In the case of CellNet and Mogrify where gene expression data from both the source and target cell-type is required, the source cell-type was fixed to fibroblast for CellNet and dermal fibroblast for Mogrify. Following this, it is possible to extract the predictions from each method for overlapping cell-types. Specifically, the sets of TF-predictions provided by each technique (15 cell-types from CellNet, 226 from JSD and 245 from Mogrify) were placed onto the cell-type ontology tree using a combination of text mining and manual curation (see Methods). summarizes this overlap of available cell conversions between each of the 3 methods. There are only 10 common cell-types between CellNet, Mogrify and JSD (skin epidermis, colon, heart, lung, liver, neuron, embryonic stem cells, female gonad, macrophage and skeletal muscle). CellNet and JSD have an additional 4 common target cell-types (kidney, B-cell, endothelial cell and haematopoietic stem cell), whereas JSD and Mogrify have 70 cell-types in common (see Fig. S1 for details). If the TF-predictions are provided by 2 or 3 methods, then the percentage of commonly predicted TFs is calculated for each cell-type and represented as a heatmap in and S1. For example, predictions for ‘M1’ and ‘M2 macrophage’ from JSD, ‘Human macrophage’ from Mogrify and ‘macrophage’ from CellNet are all annotated as ‘macrophage’ on the cell-type ontology tree. The overlap of predicted TFs between CellNet and JSD for this conversion is 33% whereas between Mogrify and both CellNet and JSD is 18%.
Figure 2. Subset of cell-type ontology tree of transdifferentiation. Cell-types available for transdifferentiation from fibroblast by each method is mapped onto the UBERON cell-type ontology tree. The pie charts indicate which of the methods provide a prediction for the cell-type on the tree. The heatmap represents the overlap of the predicted TFs between methods (i.e. Mogrify and JSD is M:J, JSD and CellNet is J:C and CellNet and Mogrify is C:M). The table provides the set of TFs predicted by each method for conversion from fibroblast. If there are more than one predictions mapped to the cell-type node, then it is denoted by * and the number of asterisks indicates the number of addition.

Analysis of the complete cell-type ontology tree shows that the mean overlap of predicted TFs by the 3 methods across common cell-types is relatively low; 18.5% of the predictions matched between Mogrify and JSD, 27.3% between JSD and CellNet and 16.7% between CellNet and Mogrify. The predictions mapping to liver had the highest overlap (an average overlap of 63.6% between methods) whereas those mapping to neuron had the lowest overlap (an average overlap of 4.7% between methods) (see Fig. S1). Since JSD and Mogrify cover more cell-types than CellNet a more detailed comparison of predicted TFs is possible. Among the 70 common cell-types between JSD and Mogrify, the most frequently predicted TFs by JSD are E2F7 and SNAI2, (predicted in 19% of cell-types) and HES1, JUNB, SOX9 by Mogrify (predicted in 16% of cell-types). In each case, the cell-types for which these TFs are being predicted are very different, and in general the predicted sets of TFs between the 2 techniques vary greatly. Furthermore, in JSD the predicted TFs appear more often to be the same among closely associated cell-types in the cell-type ontology tree when compared to Mogrify (Fig. S2B and S2C). For instance, JSD predicts IKZF1 in many of TF-sets for cell-types in the haematopoietic lineage and OLIG1 for the neural related cells, however this trend is not observed in Mogrify for the same set of cell-types. This result may be due to the fact that JSD aims to find TFs that are highly expressed in the target cell with respect of a fixed background data set. For instance, JSD's background dataset has no representation of haematopoietic related cells and as a result, this allows for IKZF1 which is a lineage rather than cell-type specific transcription factor, to be predicted for all cell-types in haematopoietic lineage. Conversely, CellNet and Mogrify have representation from across the tree in each background with the exclusion of target cell-type and as a result, TFs that are specific to cell-type rather than a lineage are detected as differentially expressed and therefore predicted (Fig. S3).
A conversion that is the subject of much investigation in the field is from fibroblast to an embryonic stem cell (ESC) like state or iPSc. This conversion was experimentally demonstrated in 2 pioneer studies using 2 different sets of TFs: OCT4, SOX2, KLF4, and MYC reported inCitation30 and OCT4, SOX2, NANOG, and LIN28A reported inCitation31. A predicted set of TFs for this conversion is provided by each of the computational techniques (). TFs that overlap with the experimentally validated studies mentioned above are listed as follows: CellNet predicts both LIN28A and NANOG; JSD predicts NANOG and OCT4 while Mogrify predicts NANOG, SOX2 and OCT4. CellNet predicts LIN28A, whereas both JSD and Mogrify do not define LIN28A as a TF. Notably, MYC and KLF4 are not predicted by any of the methods despite being classified as TFs by each of them. However, conversions to iPS cells have been possible with OCT4 and SOX2 alone.Citation32
Influence of a cell's gene expression profile on TF- prediction
Since each of the methods uses different underlying gene expression data, differences in TF-predictions can arise. JSD uses gene expression profiles quantified by microarray and Mogrify uses CAGE. To highlight the differences resulting from these 2 approaches; gene expression data from the heart, hepatocyte and embryonic stem cells are analyzed in detail (Fig. S4). For each cell-type, the gene expression profile detected by microarray and CAGE are correlated (Pearson correlation of 0.68 in heart, 0.78 in hepatocyte and 0.76 in ESC) but there are some discrepancies. For example, HIC2 in heart (Fig. S4A) and CUX2 in hepatocyte (Fig. S4B) are predicted as TFs required for cell conversion by JSD. However, Mogrify does not similarly predict these factors for transdifferentiation from fibroblast because neither TFs are expressed in the corresponding CAGE data. In contrast, both SOX2 and ZIC2 (Fig. S4C) are similarly highly expressed in CAGE and microarray, but the former is predicted only by Mogrify and the latter by CellNet for transdifferentiation to ESC. Therefore, these discrepancies can arise from the variation in underlying biological samples, the gene expression platform and the data post-processing. With these differences in mind, it may be beneficial for future methods to use a unified gene expression resource or to provide more details about the underlying samples and data collection.
TF-predictions for two well-characterized transdifferentiations
In order to further elucidate the differences between predictions from each method, we consider 2 well-studied conversions; fibroblast to cardiomyocyte-like cells and fibroblast to hepatocyte-like cells. In each case a target cell-type or tissue-type is required in order to generate a prediction from each method. In the case of cardiomyocyte-like cells heart tissue is used as the target for each method since either no cardiomyocyte sample is available or a problem with the cardiomyocyte is identified in the original publication. In the case of hepatocyte-like cells no problems are reported so we have chosen hepatocyte where available (Mogrify and JSD) and liver tissue otherwise (CellNet). The predicted TFs for each conversion for both JSD and CellNet are the top 10 ranked TFs while Mogrify predicts a set of 8 TFs for both heart tissue and hepatocyte.
Chen O, et al.Citation33 provide a detailed review on experimentally validated TFs for conversion between cardiac fibroblast and cardiac myocyte. The key transcription factors for this conversion are GATA4, MEF2C, TBX5, HAND2, MYOCB, SRF and NKX2-5. Of these, CellNet predicts one factor (TBX5), JSD 3 factors (TBX5, GATA4, NKX2-5) and Mogrify 4 factors (TBX5, GATA4, NKX2-5, MEF2C) (see for details). In order to understand the functional consequences of the TF sets chosen by each method, a gene ontology (GO) enrichment analysis was performed. This analysis looked for functional enrichment among the predicted TFs and those genes which they are known to interact with, i.e. the local network of the predicted TF set according to GeneManiaCitation34 (see Methods for details). shows the top 20 enriched biological processes (BPs) terms for each method along with their corresponding p-values. The common and unique BPs for each method are indicated along with, where possible, the identity of which TF is likely to be responsible for regulating each BP. The predictions from all 3 methods for heart tissue are enriched for genes involved in ventricular cardiac muscle tissue morphogenesis, septum morphogenesis, positive regulation of cardiac muscle cell proliferation and cardioblast differentiation. Furthermore, both JSD and Mogrify TF sets are enriched for terms involved in early development (e.g. in utero embryonic development, mesoderm formation and heart looping), which are not detected within the CellNet network. The genes that are responsible for these early developmental processes are regulated by the transcription factors HAND1, NKX2-5, GATA4 and GATA6, which are not predicted by CellNet. It has been shown that cardiac differentiation in vivo is induced by 2 master TFs, TBX5 and NKX2-5Citation35. Moreover, GATA4 and GATA6 are known to be highly important in heart formation, with the loss of both factors leading to acardia in miceCitation36 and overexpression of either resulting cardiomyocyte hypertrophy.Citation37 Likewise, HAND1 is an important regulatory protein that controls the proliferation and differentiation balance in the developing heart.Citation38 These enrichments seem to suggest that those TFs predicted by JSD and Mogrify are also involved in the coordination of heart development. In contrast, both CellNet and JSD predict a common transcription factor, ANKRD1 which is a transcriptional regulatory protein that recruits and localizes GATA4 and ERK1/2 in a sarcomeric macro-molecular complex, inducing hypertrophy.Citation39 As a result, the inclusion of this TF suggests that genes enriched for sarcomere organization and cardiac muscle contraction function are under direct regulation. Another TF predicted only by CellNet and JSD is HEY2. This TF regulates genes that are enriched for the function ‘regulation of cardiac conduction’ and it is known that the deletion of HEY2 alters myocyte action potential dynamics but does not change the function of the conduction system.Citation40 ANKRD1 and HEY2 appear to be involved in ensuring the correct function of mature heart tissue rather than having a role in development.
Figure 3. Fibroblast to heart conversion. Top 20 gene ontology biological processes (BPs) enriched in the predicted TFs network for each method (A) CellNet, (B) JSD and (C) Mogrify are given with the corresponding p-values. The method Mogrify is denoted as M, CellNet is denoted as C, and JSD is denoted as J. The common BP enriched between the methods for example CellNet, JSD and Mogrify is represented as C:J:M; and between Mogrify and CellNet is represented as M:C. The last column provides the TFs that enriches the same BP when individual TF along with the first neighbors in the network were used. The unique TFs predicted by each method are highlighted. BP terms related to regulation of transcription are written in gray and italics.
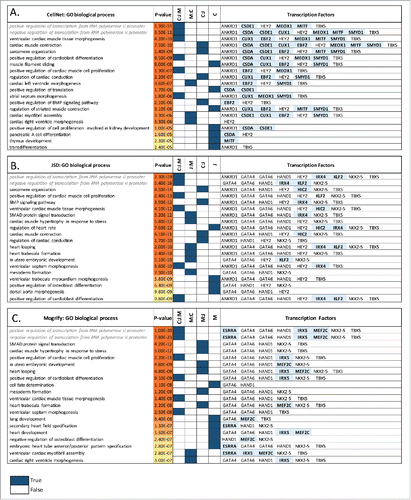
Table 1. Fibroblast to cardiomyocyte-like cells. Predicted TF sets to initiate conversion from fibroblast to heart. Those TFs shown in Bold are predicted by more than one technique and those underlined are predicted by all 3 methods.
Several experimental studiesCitation3,4,5,41,42 have used different cocktails of TFs consisting of GATA4, HNF1Α, HNF4A, CEBPA, NR1I2, FOXA2, FOXA3, CEBPB, ONECUT1, ATF5 and PROX1 to induce a conversion to hepatocyte-like cells. shows the TFs predicted by each method, and there are 4 common TFs (NR1H4, HNF4A, ATF5 and TBX5) predicted by all the methods. Furthermore, as presented in all 3 predicted TF sets regulate genes participating in steroid hormone mediated signaling, endocrine pancreas development and Notch and intracellular receptor signaling pathways. Since all 3 methods to some extent rely on the specificity of transcription factors it is not surprising that the functional enrichments are related to hepatocyte-specific processes. However, by looking at the enriched functional terms that are specific to a single method and their regulatory TF, it is possible to gain insights into the functional consequences of different transcription factor sets. The FOXA family of TFs, which are pioneer transcription factors, regulates genes enriched for functional terms related to the early embryonic development of liver, pancreas, and lungs. It has also been shown that when FOXA2/A1 is deleted, liver-specific genes were downregulated and FOXA3 could not compensate for the loss of nucleosome accessibility. In CellNet both FOXA2 and FOXA3 are predicted, however in JSD and Mogrify only FOXA2 is predicted. It has been demonstrated that only one of these factors is required for a successful conversion and as such they are redundant to each other.Citation4 A similar phenomenon can be seen elsewhere, for instance CellNet and JSD predict both NR1I2 and NR1I3 and additionally CellNet predicts both ONECUT1 and ONECUT2, but the same is not observed in Mogrify where only a single TF is predicted in each case. This is an example of Mogrify, where in the final stage it identifies and removes redundancy and provides a minimal set of TFs without affecting the predicted conversion (theoretically). The conversion of fibroblast to hepatocyte has been most robustly shown using hepatic fate conversion factors HNF1A, HNF4A, and HNF6A along with the maturation factors ATF5, PROX1, and CEBPA.Citation42 From these HNF4A and ATF5 are predicted by all of the approaches. A number of other members of the hepatic receptor family are also predicted, for instance ONECUT1 (HNF6A) is predicted by CellNet and Mogrify, but CellNet additionally predicts ONECUT2 (HNF-6-BETA). HNF4A (NR2A1) is well known master regulator of liver-specific genes and also known to have an important role in drug metabolism.Citation43 NR1H4 (FXR) is predicted by all 3 methods and is important in liver metabolism functions such as regulating bile salt synthesis and transport, and cholesterol synthesis and conversion.Citation44 Moreover, when NR1H4 interacts with PPARA (predicted by Mogrify) it is involved in fatty acid Beta-oxidation and when it interacts with NR0B2 (predicted by JSD), it decreases the bile salt synthesis by repressing CYP8B1/CYP7A1. Both CellNet and JSD predict NR1I2 and NR1I3, which have been shown to regulate CYP3A4 and this enzyme is commonly used to assess the response to xenobiotics,Citation45 a proxy for mature hepatocyte function. Experimental validation would be required to understand the effect of using these different nuclear receptor genes in this transdifferentiation, but it seems likely given the diversity of liver function that different populations of cells might require different sets of TFs in order to facilitate specific functions within the liver.
Figure 4. Fibroblast to hepatocyte conversion. Top 20 gene ontology biological processes (BPs) enriched in the predicted TFs network for each method (A) CellNet, (B) JSD and (C) Mogrify are given with the corresponding p-values. The method Mogrify is denoted as M, CellNet is denoted as C, and JSD is denoted as J. The common BP enriched between the methods for example CellNet, JSD and Mogrify is represented as C:J:M; and between Mogrify and CellNet is represented as M:C. The last column provides the TFs that enriches the same BP when individual TF along with the first neighbors in the network were used. The unique TFs predicted by each method are highlighted. BP terms related to regulation of transcription are written in gray and italics.
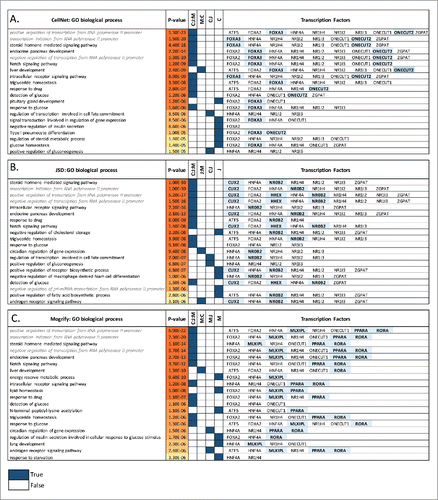
Table 2. Fibroblast to hepatocytes-like cells. Predicted TF sets to initiate conversion from fibroblast to hepatocyte. Those TFs shown in Bold are predicted by more than one technique and those underlined are predicted by all 3 methods.
Discussion
The development of data-driven approaches to identify TFs cocktail for transdifferentiation is a major step forward in the field. The transition from trial-and-error experimental approaches to systematically identified TF sets not only increases the rate at which new transdifferentiation can be discovered but will allow a clearer understanding of how this process is controlled biologically. However, there are a many issues that still remain and which affect each of the existing prediction algorithm. These issues along with possible enhancements that can be made are as follows.
First and foremost is that any data-driven approach is limited in both depth and quality by the availability of the data. Predictions for many scientifically and clinically interesting cell- and tissue-types are not available in any of the existing techniques owing to the fact of limited availability of expression data in the required format. Even though JSD and Mogrify try to provide predictions for a broader set of cell-types (i.e., 233 and 272 cell-types respectively) when compared to CellNet (16 cell-types), they still do not provide many additional clinically relevant cell-types. In this sense, it is important that as the techniques are now available the focus can shift to a more extensive acquisition of high-quality data for more cell-types.
Currently very little is known about the effect of variability between cellular sources of data. The effect of the data being collected from different individuals or at different development stages could result in very different predicted transcription factors. Likewise, as tissues are made of heterogeneous cell-types, the predictions for tissues like liver or heart is less informative when compared to pure cell-populations like cardiomyocyte, cardiac fibroblast or hepatocyte cardiomyocytes, cardiac fibroblasts or hepatocytes. Recently, single-cell RNA-Seq expression profile of transcriptome states during the lineage differentiation (such as fibroblast to neuronCitation46) are being used to gain deeper insights in the transdifferentiation process and this information can be utilised to improve the precision of prediction.
For transdifferentiation the starting cell-type is essential to determine the factors required for conversion as cells from a different lineage may require a different set of TFs to result in the same target cell. For example, Wei R, et al.Citation47 provide a detailed review of pancreatic ß cell generation by ectopic expression of TFs from different starting cell-types such as keratinocytes (PDX1, NGN3 and NEUROD), neural cells (GLUT2, GK, NGN3 and NEUROD) and biliary cells (PDX1, NGN3 and MAFA). CellNet and Mogrify appropriately take into account the starting cell-type for predictions while JSD fails to attempt to account for this variation.
We showed that the underlying differences in the TFs-predictions from each method can arise due to the cell samples used, gene expression quantification methods as well as the algorithm. JSD tends to predict the same TFs for closely associated cell-types but the extent to which this is due to the background selection or the algorithm is unclear. The problem of how to select the best background with which to compare the expression profile of a target cell-type is not trivial, and the approaches taken by each of the 3 methods have both advantages and disadvantages.
In the case of fibroblast to cardiomyocyte-like and hepatocyte-like cell, JSD and Mogrify tend to predict TFs that are more involved in the developmental process in contrast to CellNet. Additionally, JSD and CellNet tend to predict functionally redundant genes while this is not observed in Mogrify and might be due to its exclusion of TFs with overlapping regulatory influence.
The availability of cell-type specific data on TF interactions means that the networks used by these approaches are created either by correlation of expression (CellNet) or via incorporating existing resources (Mogrify). These approaches are likely to introduce many false positive (FP) connections which will undoubtedly have a knock-on effect to the quality of the predicted TF set. With approaches such as ChiP-seq and ATAC-seq now facilitating high-throughput profiling of TF binding events, it should be possible to reduce the number of FPs by identifying and removing the predicted edges for which there is no experimental support.
Finally, for the purpose of cell conversion there is no unified definition of a TF and different methods use different definitions to make their predictions. For instance, an important factor like LIN28A (used for transdifferentiation of fibroblast to iPSc) is considered as TF in CellNet but not in either Mogrify or JSD. A unified definition of TF or a ‘gold-standard’ repository would allow future techniques to be more consistent in the breadth of factors that can be predicted.
Beyond using TFs, Cao N, et al.Citation48 recently achieved a chemically induced transdifferentiation from human fibroblast to cardiomyocytes using a combination of 9 compounds consisting of small molecules and growth factors. These chemically induced cardiomyocytes (ciCMs) were similar to human cardiomyocytes based on transcriptome, epigenetic and electrophysiological properties. This discovery indicates that the future of transdifferentiations may not rely on TF over-expression alone, and other ectopic factors such as growth factors, chemicalsCitation48,49,50 and epigenetic remodellersCitation51 should be considered as candidates for enhanced cell conversion. As a result of this diversification in reprogramming stimuli, it will be important that computational methods focus on integrating these factors in their predictions. Finally and above all, it is still the case that the number of successful conversions is still relatively low. Hence it is important that as a field that more conversions are attempted, and details of both successful and unsuccessful conversions are reported so that the approach of identifying the correct factors for reprogramming can be refined.
Methods
Cell-types ontology tree construction
In order to compare the TFs predicted for a given conversion by each method, the cell-types have been mapped to a common ontology tree representing all the cell-types available in the 3 methods.
In the case of CellNet and Mogrify, the software makes predictions based on both source and target cell-type information. Therefore, for these two methods a commonly used source cell-type was selected, fibroblast for CellNet and dermal fibroblast for Mogrify. CellNet required an external microarray data of the source cell-type, hence we used fibroblast expression data (GSM372142, GSM372144 and GSM372146) in GSE14897Citation52 experiment obtained from GEO. Given the selected source cell-type, the available target cell-types for conversion consist of 15 cell-types for CellNet and 272 cell-types for Mogrify. As the TFs predictions provided by JSD are generated irrespective of the source cell, all 233 cell-types from their published atlas including fibroblast are used for comparison.
The predicted TF sets for JSD and CellNet are considered to be the top 10 ranked TFs. Mogrify predicts a non-redundant TF set for each conversion varying from 1 to 8 TFs. To make the predictions between methods comparable, firstly all the predicted TF-gene symbols were converted to their corresponding HUGOCitation53 approved gene symbol. Secondly, to reduce the multiple mapping we used only cell-types from adult samples and also removed intermediate transdifferentiation stage cell-types which further led to 226 cell-types in JSD and 245 cell-types in Mogrify.
As the cell-type names from each method were different and did not follow any standard ontology, we tried to unify the cell-types from the three methods in terms UBERON extended ontologyCitation29 which comprises of the Uber anatomy Ontology (UBERON), Cell Type (CL) and Gene Ontology (GO). From the UBERON extended ontology, all the cell-type nodes with relationship (edges) of type ‘is a’, ‘part of’ and ‘derived from’ were selected to form a directed ontology network made of 17,822 nodes and 41,051 edges. Each node in this network is represented by an ontology ID and its respective approved name, synonymous names and any obsolete terms. To find the best-matched ontology ID on the network for a given cell-type, we text matched the cell-type name given by the method with every ontology network node's attribute names (approved, synonymous and obsolete terms). The ontology ID was then assigned to each cell-type based on the highest common words in the names. Following this, a manual curation of the ID association for some of the cell-types was performed to ensure that the match was correct. The result of this was a specific ontology ID on the cell-type ontology network associated to every cell-type from each of the methods. Next to reduce the complexity of the network the 320 nodes that contained at least one cell-type and their connecting nodes were selected from the cell-type ontology network. Finally, the intermediate nodes with an in-degree of one and an out-degree of one were removed. This led to a final minimal ontology network with 1,428 nodes and 3,777 edges containing all the 320 unique target cell-types.
With the aim to calculate the phylogenetic distances and cluster the cell-types, we converted this network into a neighbour-joining tree based on the shortest undirected path between every pair of target cell-type available in the methods. NetworkXCitation54, which is a python module, was used to manipulate the ontology network and APE, which is an R packageCitation55, was used to generate the phylogenetic ontology tree of cell-types. Table S1 provides the cell-type name given by the methods, their associated ontology ID on the tree and TFs predicted for all conversions from fibroblast. The cell-type tree along with TFs predicted shown in , S2, S3 were visualised in iToLCitation56
Functional enrichment analysis
For the transdifferentiation case studies (fibroblast to cardiomyocyte-like and hepatocyte-like cell-type), in order to annotate the biological functions of the genes that the TFs predicted by each method regulate, GeneManiaCitation34 was used to generate a network around each set of TFs as this technique had a minimal bias towards any of the methods. This software provides genes that interact with a set of TFs based on the interaction information obtained from gene co-expression from GEOdbCitation20, physical and genetic interaction from BioGridCitation57, pathway interaction from ReactomeCitation58 and BioCyc, shared protein domains and co-localization. The set of TFs predicted is extended to a network by adding 100 genes based on (1) known gene-gene interactions or co-expression (etc., see above) and (2) to favour a common biological function. This resulted in an undirected network with 110 genes for CellNet and JSD, and 108 genes for Mogrify in both the cardiomyocyte-like and hepatocyte-like case studies.
In order to identify which biological functions each TFs set is involved in, gene ontology enrichment analysis was performed. For each method, the gene ontology biological processes (BPs) enriched by the TFs and genes network were identified using topGOCitation59 package in R. The top 20 most significant BPs based on Fisher exact test p-value and the eliminated option to reduce redundancy in GO terms were used for further analysis. To determine whether a specific TF is responsible for the network's BP enrichment, a subnetwork consisting of the TF and its first gene neighbors is extracted from the larger network and analysed for functional enrichment. The TF is considered associated with the network's BP enrichment if the larger network's BP is found in the top 100 BPs enriched by the TF-neighbors subnetwork. and show the top 20 network enriched biological process with a set of TFs associated.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1238119_Supplemental_Material.zip
Download Zip (3.7 MB)Related Research Data
References
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987; 51:987-1000; PMID:3690668; http://dx.doi.org/10.1016/0092-8674(87)90585-X
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663-76; PMID:16904174; http://dx.doi.org/10.1016/j.cell.2006.07.024
- Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, Hu Y, Wang X, Hui L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 2011; 475:386-9; PMID:21562492; http://dx.doi.org/10.1038/nature10116
- Sekiya S, Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 2011; 475:390-3; PMID:21716291; http://dx.doi.org/10.1038/nature10263
- Kogiso T, Nagahara H, Otsuka M, Shiratori K, Dowdy SF. Transdifferentiation of human fibroblasts into hepatocyte-like cells by defined transcriptional factors. Hepatol Int 2013; 7:937-44; PMID:26201932; http://dx.doi.org/10.1007/s12072-013-9432-5
- Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010; 142:375-86; PMID:20691899; http://dx.doi.org/10.1016/j.cell.2010.07.002
- Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012; 485:599-604; PMID:22660318; http://dx.doi.org/10.1038/nature11139
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J, Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012; 485:593-8; PMID:22522929; http://dx.doi.org/10.1038/nature11044
- Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol 2016; 17:183-93; PMID:26883003; http://dx.doi.org/10.1038/nrm.2016.8
- Sadahiro T, Yamanaka S, Ieda M. Direct cardiac reprogramming: Progress and challenges in basic biology and clinical applications. Circ. Res. 2015; 116:1378-91.
- Tsunemoto RK, Eade KT, Blanchard JW, Baldwin KK. Forward engineering neuronal diversity using direct reprogramming. EMBO J 2015; 34:1445-55; PMID:25908841; http://dx.doi.org/10.15252/embj.201591402
- Heinäniemi M, Nykter M, Kramer R, Wienecke-Baldacchino A, Sinkkonen L, Zhou JX, Kreisberg R, Kauffman S a, Huang S, Shmulevich I. Gene-pair expression signatures reveal lineage control. Nat Methods 2013; 10:577-83; http://dx.doi.org/10.1038/nmeth.2445
- Lang AH, Li H, Collins JJ, Mehta P. Epigenetic landscapes explain partially reprogrammed cells and identify key reprogramming genes. PLoS Comput Biol 2014; 10:e1003734; PMID:25122086; http://dx.doi.org/10.1371/journal.pcbi.1003734
- Del Sol ICA. A general strategy for cellular reprogramming: The importance of transcription factor cross-repression. Stem Cells 2013; 31:2127-35; PMID:23873656; http://dx.doi.org/10.1002/stem.1473
- Davis FP, Eddy SR. Transcription factors that convert adult cell identity are differentially polycomb repressed. PLoS One 2013; 8:1-8.
- D'Alessio AC, Fan ZP, Wert KJ, Baranov P, Cohen MA, Saini JS, Cohick E, Charniga C, Dadon D, Hannett NM, et al. A systematic approach to identify candidate transcription factors that control cell identity. Stem Cell Reports 2015; 5:763–75.
- Cahan P, Li H, Morris SA, Lummertz da Rocha E, Daley GQ, Collins JJ. Cellnet: network biology applied to stem cell engineering. Cell 2014; 158:903-15.
- Rackham OJL, Firas J, Fang H, Oates ME, Holmes ML, Knaupp AS, Suzuki H, Nefzger CM, Daub CO, Shin JW, et al. A predictive computational framework for direct reprogramming between human cell types. Nat Genet 2016; 48(3):331–5; PMID:26711105
- Bian Q, Cahan P. Computational tools for stem cell biology. Trends Biotechnol 2016; in press.
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: Archive for functional genomics data sets - Update. Nucleic Acids Res 2013; 41:D991-5; PMID:23193258; http://dx.doi.org/10.1093/nar/gks1193
- Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S, Abugessaisa I, Fukuda S, Hori F, Ishikawa-Kato S, et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol 2015; 16:22; PMID:25723102; http://dx.doi.org/10.1186/s13059-014-0560-6
- Draghici S, Khatri P, Eklund AC, Szallasi Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006; 22:101-9; PMID:16380191; http://dx.doi.org/10.1016/j.tig.2005.12.005
- Rosvall M, Bergstrom CT. An information-theoretic framework for resolving community structure in complex networks. Proc Natl Acad Sci U S A 2007; 104:7327-31; PMID:17452639; http://dx.doi.org/10.1073/pnas.0611034104
- Efron B, Tibshirani R. ON TESTING THE SIGNIFICANCE OF SETS OF GENES. Ann Appl Stat 2007; 1:107-29; http://dx.doi.org/10.1214/07-AOAS101
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106; PMID:20979621; http://dx.doi.org/10.1186/gb-2010-11-10-r106
- Addis RC, Ifkovits JL, Pinto F, Kellam LD, Esteso P, Rentschler S, Christoforou N, Epstein JA, Gearhart JD. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J Mol Cell Cardiol 2013; 60:97-106; PMID:23591016; http://dx.doi.org/10.1016/j.yjmcc.2013.04.004
- Consortium TF, Omics R. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat Genet 2009; 41:553-63; PMID:19377474; http://dx.doi.org/10.1038/ng.375
- Wingender E, Schoeps T, Dönitz J. TFClass: an expandable hierarchical classification of human transcription factors. Nucleic Acids Res 2013; 41:D165-70; PMID:23180794; http://dx.doi.org/10.1093/nar/gks1123
- Haendel MA, Balhoff JP, Bastian FB, Blackburn DC, Blake JA, Bradford Y, Comte A, Dahdul WM, Dececchi TA, Druzinsky RE, et al. Unification of multi-species vertebrate anatomy ontologies for comparative biology in Uberon. J Biomed Semantics 2014; 5:21; PMID:25009735; http://dx.doi.org/10.1186/2041-1480-5-21
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131:861-72; PMID:18035408; http://dx.doi.org/10.1016/j.cell.2007.11.019
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318:1917-20; PMID:18029452; http://dx.doi.org/10.1126/science.1151526
- Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, Melton DA. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 2008; 26:1269-75; PMID:18849973; http://dx.doi.org/10.1038/nbt.1502
- Chen O, Qian L. Direct Cardiac Reprogramming: Advances in Cardiac Regeneration. Biomed Res Int 2015; 2015:1-8.
- Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 2010; 38; PMID:20576703; http://dx.doi.org/10.1093/nar/gkq537
- Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet 2001; 28:276-80; PMID:11431700; http://dx.doi.org/10.1038/90123
- Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol 2008; 317:614-9; PMID:18400219; http://dx.doi.org/10.1016/j.ydbio.2008.03.013
- Liang Q, De Windt LJ, Witt SA, Kimball TR, Markham BE, Molkentin JD. The Transcription Factors GATA4 and GATA6 Regulate Cardiomyocyte Hypertrophy in Vitro and in Vivo. J Biol Chem 2001; 276:30245-53; PMID:11356841; http://dx.doi.org/10.1074/jbc.M102174200
- Risebro CA, Smart N, Dupays L, Breckenridge R, Mohun TJ, Riley PR. Hand1 regulates cardiomyocyte proliferation versus differentiation in the developing heart. Development 2006; 133:4595-606; PMID:17050624; http://dx.doi.org/10.1242/dev.02625
- Zhong L, Chiusa M, Cadar AG, Lin A, Samaras S, Davidson JM, Lim CC. Targeted inhibition of ANKRD1 disrupts sarcomeric ERK-GATA4 signal transduction and abrogates phenylephrine-induced cardiomyocyte hypertrophy. Cardiovasc Res 2015; 106:261-71; PMID:25770146; http://dx.doi.org/10.1093/cvr/cvv108
- Hartman ME, Liu Y, Zhu WZ, Chien WM, Weldy CS, Fishman GI, Laflamme MA, Chin MT. Myocardial deletion of transcription factor CHF1/Hey2 results in altered myocyte action potential and mild conduction system expansion but does not alter conduction system function or promote spontaneous arrhythmias. FASEB J 2014; 28:3007-15; PMID:24687990; http://dx.doi.org/10.1096/fj.14-251728
- Pournasr B, Asghari-Vostikolaee MH, Baharvand H. Transcription factor-mediated reprograming of fibroblasts to hepatocyte-like cells. Eur J Cell Biol 2015; 94:603-10; PMID:26561000; http://dx.doi.org/10.1016/j.ejcb.2015.10.003
- Du Y, Wang J, Jia J, Song N, Xiang C, Xu J, Hou Z, Su X, Liu B, Jiang T, et al. Human hepatocytes with drug metabolic function induced from fibroblasts by lineage reprogramming. Cell Stem Cell 2014; 14:394-403; PMID:24582926; http://dx.doi.org/10.1016/j.stem.2014.01.008
- Firas J, Liu X, Lim SM, Polo JM. Transcription factor-mediated reprogramming: epigenetics and therapeutic potential. Immunol Cell Biol 2015; 93:284-9; PMID:25643615; http://dx.doi.org/10.1038/icb.2015.5
- Zimmer V, Liebe R, Lammert F. Nuclear receptor variants in liver disease. Digestive Diseases 2015; 33(3):415-9.
- Tirona RG, Lee W, Leake BF, Lan L-B, Cline CB, Lamba V, Parviz F, Duncan SA, Inoue Y, Gonzalez FJ, et al. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med 2003; 9:220-4; PMID:12514743; http://dx.doi.org/10.1038/nm815
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SAM, Sim S, Neff NF, Skotheim JM, Wernig M, et al. Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 2016; 534:391-5; PMID:27281220; http://dx.doi.org/10.1038/nature18323
- Wei R, Hong T. Lineage Reprogramming: A Promising Road for Pancreatic β Cell Regeneration. Trends Endocrinol. Metab. 2016; 27:163-76.
- Cao N, Huang Y, Zheng J, Spencer CI, Zhang Y, Fu J-D, Nie B, Xie M, Zhang M, Wang H, et al. Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science 2016; 352(6290):1216-20
- Fu Y, Huang C, Xu X, Gu H, Ye Y, Jiang C, Qiu Z, Xie X. Direct reprogramming of mouse fibroblasts into cardiomyocytes with chemical cocktails. Cell Res 2015; 25:1013-24; PMID:26292833; http://dx.doi.org/10.1038/cr.2015.99
- Zhang M, Lin Y, Sun YJ, Li K, Zhang M, Lin Y, Sun YJ, Zhu S, Zheng J, Liu K. Pharmacological reprogramming of fibroblasts into neural stem cells by signaling-directed transcriptional activation article pharmacological reprogramming of fibroblasts into neural stem cells by signaling-directed transcriptional activation. Cell Stem Cell 2016; 18:653–67.
- Katz LS, Geras-Raaka E, Gershengorn MC. Reprogramming adult human dermal fibroblasts to islet-like cells by epigenetic modification coupled to transcription factor modulation. Stem Cells Dev 2013; 22:2551-60; PMID:23627894; http://dx.doi.org/10.1089/scd.2013.0134
- Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010; 51:297-305; PMID:19998274; http://dx.doi.org/10.1002/hep.23354
- Povey S, Lovering R, Bruford E, Wright M, Lush M, Wain H. The HUGO Gene Nomenclature Committee (HGNC). Hum Genet 2001; 109:678-80; PMID:11810281; http://dx.doi.org/10.1007/s00439-001-0615-0
- Hagberg AA, Schult DA, Swart PJ. Exploring network structure, dynamics, and function using NetworkX. Network 2008; 836:11-5.
- Paradis E, Claude J, Strimmer K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004; 20:289-90; PMID:14734327; http://dx.doi.org/10.1093/bioinformatics/btg412
- Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 2016; 44:W242–5.
- Breitkreutz BJ, Stark C, Reguly T, Boucher L, Breitkreutz A, Livstone M, Oughtred R, Lackner DH, Bähler J, Wood V, et al. The BioGRID interaction Database: 2008 update. Nucleic Acids Res 2008; 36:D637-40; PMID:18000002
- Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, Caudy M, Garapati P, Gillespie M, Kamdar MR, et al. The Reactome pathway knowledgebase. Nucleic Acids Res 2014; 42:D472-7; http://dx.doi.org/10.1093/nar/gkt1102
- Alexa A, Rahnenfuhrer J. topGO: topGO: Enrichment analysis for Gene Ontology. R package version 2.18.0. October 2010;