
ABSTRACT
Anti-silencing function 1 (ASF1) is a histone H3-H4 chaperone involved in DNA replication and repair, and transcriptional regulation. Here, we identify ASF1B, the mammalian paralog to ASF1, as a proliferation-inducing histone chaperone in human β-cells. Overexpression of ASF1B led to distinct transcriptional signatures consistent with increased cellular proliferation and reduced cellular death. Using multiple methods of monitoring proliferation and mitotic progression, we show that overexpression of ASF1B is sufficient to induce human β-cell proliferation. Co-expression of histone H3.3 further augmented β-cell proliferation, whereas suppression of endogenous H3.3 attenuated the stimulatory effect of ASF1B. Using the histone binding-deficient mutant of ASF1B (V94R), we show that histone binding to ASF1B is required for the induction of β-cell proliferation. In contrast to H3.3, overexpression of histone H3 variants H3.1 and H3.2 did not have an impact on ASF1B-mediated induction of proliferation. Our findings reveal a novel role of ASF1B in human β-cell replication and show that ASF1B and histone H3.3A synergistically stimulate human β-cell proliferation.
Introduction
The replicative capacity of adult β-cells is very limited, and sharply declines with age. In adult humans, <0.5% of the β-cells replicate, vs. ∼3% during neo- or perinatal stages.Citation1 Similarly, in older mice, β-cells replicate at <0.2%, vs. ∼2.5% in young mice.Citation2,3 However, impaired glucose tolerance, pregnancy, dietary challenge, and peripheral insulin resistance have been shown to induce β-cell expansion.Citation4-6 This metabolic stress-induced increase in β-cell mass most likely reflects the replication of existing β-cells, rather than differentiation of stem cells or other progenitor cell types.Citation5,7 Further, it suggests that the physiological triggers that stimulate β-cell proliferation are present and functional in adult humans. Failure to compensate with increased β-cell proliferation in response to these physiological stimuli leads to a loss in functional β-cell mass and ultimately, type 2 diabetes. Therefore, a major focus in the field is to identify these physiological triggers and determine if they can be exploited to enhance β-cell proliferation and restore β-cell mass in diabetes.Citation1,8,9
Several signaling pathways have been linked to β-cell proliferation in rodents. However, much less is known about the control of β-cell proliferation in human islets.Citation8-10 Glucokinase signaling, carbohydrate response element-binding protein (ChREBP), nuclear factor of activated T-cells (NFAT), platelet-derived growth factor (PDGF), CDK4 and TCF7L2 have all been reported to stimulate human β-cell proliferation.Citation11-16 In addition, Cdk6 and other regulators of the G1/S transition have been shown to promote proliferation in human β-cells.Citation17 These observations, coupled with the high levels of expression of numerous key cell-cycle molecules in human islets, indicate that it is likely a failure to activate the cell cycle in response to upstream signals that impairs human β-cell proliferation under physiological conditions.
Histones play a critical role in chromatin regulation and gene expression.Citation18 Histone chaperones orchestrate nucleosome assembly in several DNA-dependent processes. However, their role in cell proliferation has only recently been investigated.Citation19,20 Anti-silencing function 1 (ASF1) is a conserved histone chaperone. Originally identified in budding yeast, ASF1 regulates transcription by de-repressing (i.e., anti-silencing) silent mating-type loci.Citation21 Its function as a transcriptional regulator is evolutionarily conserved and has been reported in several organisms.Citation22-27 In addition, ASF1 chaperones have also been reported to play crucial roles in regulation of cell cycle progression in yeast strains, C. elegans and mammalian cell lines.Citation28-31
In mammals, ASF1 exists as 2 paralogs, ASF1A and ASF1B,Citation31 that share ∼70% sequence identity, but are not functionally equivalent. Studies on their functional specialization suggest that ASF1A also participates in pathways that are not exclusive to S-phase (including DNA-damage repair pathways), whereas ASF1B is involved in cell proliferation.Citation29,32 The functional specification between ASF1 paralogs is also suggested by their distinct expression pattern. In mammals, ASF1A is ubiquitously expressed, whereas ASF1B is limited to proliferating tissue, and is greatly reduced in terminally differentiated and quiescent cells.Citation33 ASF1 paralogs interact with secondary chaperones, chromatin assembly factor 1 (CAF-1) and histone regulator A (HIRA), which bind to the histone variants H3.1 and H3.3, respectively. In the replication-coupled (RC) pathway, H3.1 and H3.2 are incorporated into nucleosomes during S-phase of the cell cycle. In contrast, H3.3 is incorporated during replication-independent (RI) nucleosome assembly.Citation34 These interactions are well-established for ASF1A,Citation35 however little is known about interactions of ASF1B and its preferential involvement in RC vs. RI nucleosome assembly. Interestingly, among ASF1 paralogs, secondary chaperones and histones required for RC or RI nucleosome assembly, only the expression of ASF1B gene is greatly reduced in adult human β-cells.
We previously reported that obesity (Leptinob/ob) induced the expression of a module of cell cycle regulatory genes in islets from diabetes-resistant B6, but not diabetes-susceptible BTBR mice; i.e., induction of the cell cycle genes correlated with resistance to diabetes.Citation36 β-cell proliferation showed the same trend; obesity-dependent induction in B6 islets, but not BTBR islets. In addition to core cell cycle genes (e.g., cyclins, Cdk’s, E2Fs, etc.), the islet gene module also contained genes not known to regulate cell cycle progression in islets, including ASF1B. We hypothesized that one or more of these differentially regulated genes may be novel mediators of β-cell proliferation in response to a physiological trigger.
In this study, we examine the role of ASF1B in human β-cell proliferation. We report that overexpression of ASF1B significantly induces human β-cell proliferation by promoting S-phase entry and mitotic progression. Importantly, we show that ASF1B requires histone H3.3 to mediate its regulatory effects. These findings identify ASF1B-histone H3.3 as a novel gene-histone regulatory axis important for human β-cell proliferation.
Results
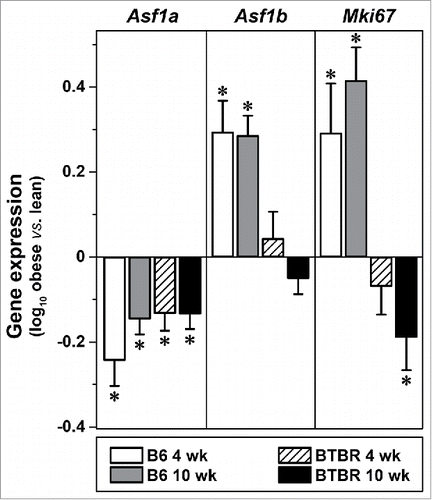
We investigated Asf1b expression in pancreatic islets of our BTBR-ob/ob mouse model of obesity-induced type 2 diabetes, and in non-diabetic control mice, B6-ob/ob. Asf1b was suppressed in response to age in both B6 and BTBR islets,Citation36 consistent with other studies reporting a decline in β-cell proliferation with age.Citation2,3 In addition to an age-dependent suppression, the expression of Asf1b was induced by obesity in B6, but not BTBR islets. Interestingly, the lack of obesity regulation in BTBR mice occurred prior to the onset of diabetes (4 weeks), suggesting that the lack of Asf1b induction is not a consequence of diabetes.Citation36 In parallel with Asf1b, obesity induced the expression of Mki67 only in B6 mice. In contrast to Asf1b, obesity resulted in the suppression of Asf1a equally in B6 and BTBR islets ().
Figure 1. Expression of Asf1b, but not Asf1a, strongly correlates with Mki67 in mouse islets. Obesity-dependent changes in the expression of Asf1a, Asf1b, and Mki67 in islets from B6 or BTBR mice at 4 or 10 weeks of age (male, N = 5 each). Gene expression values are log10-transformed ratios of individual measurements vs. a strain-specific reference pool.Citation36 *, P ≤ 0.05 for obese (Leptinob/ob) vs. lean.
ASF1B transcriptionally regulates cellular proliferation and apoptotic genes
ASF1 proteins have been implicated in transcriptional gene regulation in mouse, yeast and Drosophila.Citation37-39 More specifically, ASF1B has been identified as a cellular interaction partner of transcriptional coactivator HCF-1 in yeast, which suggests that ASF1B may also be involved in transcriptional regulation.Citation40 We used RNA sequencing to identify ASF1B-responsive genes in human pancreatic islets. Human islets were transduced with adenovirus containing ASF1B (Ad-ASF1B) and either GFP (Ad-GFP) or LacZ (Ad-LacZ), as negative controls. After 48 hr, whole islet RNA was isolated and used for RNA-sequencing. In response to ASF1B overexpression, ∼460 genes were differentially expressed (DE) ( and Table S2). Among the DE genes, 286 were induced, while 179 were repressed.
Figure 2. ASF1B modulates genes regulating cell proliferation and apoptosis in human β-cells. (A) Heat map illustrating differentially expressed (DE) genes in response to Ad-ASF1B overexpression in 10 human islet samples. Color corresponds to Z-score (decreased, blue; increased, red) relative to Ad-GFP treated islets. (B) Gene Ontology (GO) analysis of DE genes in response to ASF1B. Pie charts represent the number of genes for significant categories (Z > 2.0). A complete listing of all enriched terms is provided in Table S3.
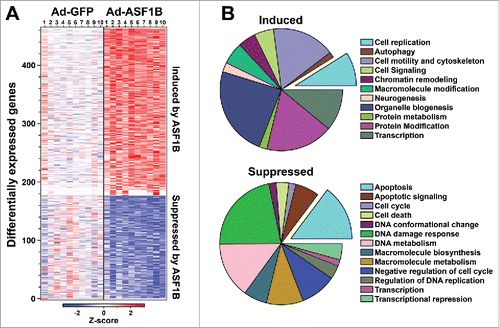
ASF1B interacts with histones H3.1 and H3.3, suggesting its possible role in both RC and RI histone management.Citation19 Its interaction with the B-domain of secondary chaperone HIRACitation41 suggests that ASF1B may also be involved in RI histone exchange leading to transcriptional gene regulation. Therefore, some of the DE genes are likely secondary transcriptional targets of ASF1B. Several genes known to be involved in cellular proliferation were induced, including CHEK1, CDKN1A, PCNA, as well as the MCM and histone family genes. Expression of several genes associated with transcriptional regulation were also induced, including MECP2, MED21, GRHL3, SMAD4, POU4F1, HES6, CUX1, and many of the ZNF family transcription factors. Among repressed genes, a significant cluster consisted of genes associated with apoptosis (BAX, BCL2L13, TCF7L2, TGM2, FAS, CUL5, MDM2 and TNFRSF10 family). In addition to the expected genes, many not known to play a role in cellular proliferation or survival were also differentially regulated, e.g., GPR87, TRIAP1, TRIM40, NHLH2, NR1D1, ZNF556 and ZNF367 (Table S2). We confirmed the gene expression results from the RNA-sequencing, by qRT-PCR for several cell cycle genes, as well as other genes that were highly induced or repressed in response to ASF1B-overexpression (Fig. S1).
To identify functional categories of genes regulated by ASF1B, we performed gene ontology (GO) analysis. There was a significant enrichment for genes involved in cell cycle regulation and mitotic progression, cell morphogenesis, chromatin and histone modifications ( and Table S3). Interestingly, among the repressed genes, a majority were enriched in apoptosis and pathways related to cell death (e.g. the p53 signaling pathway). In summary, ASF1B leads to the de-repression of several cell cycle genes, and eventually induction in cellular proliferation that could explain its strong correlation with MKi67 expression in obese non-diabetic mice.
ASF1B overexpression induces proliferation of human β-cells
Human islet cells are primarily post-mitotic, undergoing very little replication when maintained in culture. The induction of cell cycle regulatory genes in response to ASF1B suggested that the cell cycle was activated, leading to increased cellular proliferation. To directly evaluate a role for ASF1B in islet cell proliferation, we transduced intact human islets with Ad-ASF1B for 12 hr, followed by a post-transduction recovery period of 24 hr, during which islets were pulsed with BrdU to mark actively proliferating cells. We observed a significant increase in ASF1B protein abundance in Ad-ASF1B treated islets by immunohistochemistry (Fig. S2A) and by western blot analysis (, top panel). Beta-cells were identified by co-staining transduced islet sections for insulin (; see also Fig. S3 for enlarged images). The number of BrdU+ β-cells was >4-fold higher in islets overexpressing ASF1B than in control islets. Ad-ASF1B transduction effectively induced expression of ASF1B in both α-cells and β-cells, however, it did not significantly induce proliferation of non-β-cells, suggesting its mitogenic influence is specific to β-cells (Fig. S2B, C). To provide additional evidence that ASF1B promotes cellular division in human islets, we measured the incorporation of [3H]-thymidine into newly-synthesized DNA in Ad-ASF1B vs. Ad-GFP treated islets. ASF1B induced ∼1.5-fold more [3H]-thymidine incorporation (∼120 CPM/µg protein) vs. the Ad-GFP control virus (70 CPM/µg protein) in human islets (). ASF1B did not affect glucose-stimulated insulin secretion (GSIS) or insulin content, suggesting that overexpression of ASF1B was not detrimental for β-cell function (Fig. S4).
Figure 3. ASF1B overexpression induces proliferation of human β-cells. (A) Representative confocal images (60X) of intact human islets (N = 400) treated with Ad-LacZ or Ad-ASF1B, and labeled with BrdU (white) to mark proliferating cells (48 h). β-cells are identified by insulin (red), α-cells by glucagon (green) and nuclei by DAPI (blue). (B) [3H]-thymidine incorporation into human islet DNA measured 48 h after Ad-LacZ or Ad-ASF1B treatment (N = 10). (C) Flow cytometry quantitation of the % cells in G0/G1, S and G2/M phases 48 h after Ad-LacZ or Ad-ASF1B treatment of dispersed human islet cells (N = 4). (D) Representative histograms of FACS-analyzed cell cycle profiles of disrupted human islets. Data represent means ± SEM, N = 3. *, P ≤ 0.05. All comparisons are for Ad-LacZ or Ad-GFP vs. Ad-ASF1B.
![Figure 3. ASF1B overexpression induces proliferation of human β-cells. (A) Representative confocal images (60X) of intact human islets (N = 400) treated with Ad-LacZ or Ad-ASF1B, and labeled with BrdU (white) to mark proliferating cells (48 h). β-cells are identified by insulin (red), α-cells by glucagon (green) and nuclei by DAPI (blue). (B) [3H]-thymidine incorporation into human islet DNA measured 48 h after Ad-LacZ or Ad-ASF1B treatment (N = 10). (C) Flow cytometry quantitation of the % cells in G0/G1, S and G2/M phases 48 h after Ad-LacZ or Ad-ASF1B treatment of dispersed human islet cells (N = 4). (D) Representative histograms of FACS-analyzed cell cycle profiles of disrupted human islets. Data represent means ± SEM, N = 3. *, P ≤ 0.05. All comparisons are for Ad-LacZ or Ad-GFP vs. Ad-ASF1B.](/cms/asset/b4851929-2998-40c4-8f73-64dfba114e26/kccy_a_1241914_f0003_c.gif)
Our transcriptomic data showed a significant induction in genes that are known to regulate the G1/S transition of the cell cycle. To provide direct evidence that ASF1B regulates the G1/S checkpoint of the cell cycle, we quantified islet cellular DNA content by flow cytometry (). Compared to control islets, ASF1B overexpression resulted in a >20-fold increase in the population of cells in S phase (from ∼0.5% to ∼13%). ASF1B induced a corresponding decrease in the cellular population in the G1 phase; 97% in control vs. 77% in ASF1B-transduced cells (). In summary, our multiple measures of cellular proliferation (BrdU and [3H]-thymidine incorporation into islet DNA, and FACS-based DNA content in different phases of the cell cycle) provide strong evidence that ASF1B promotes proliferation of human β-cells.
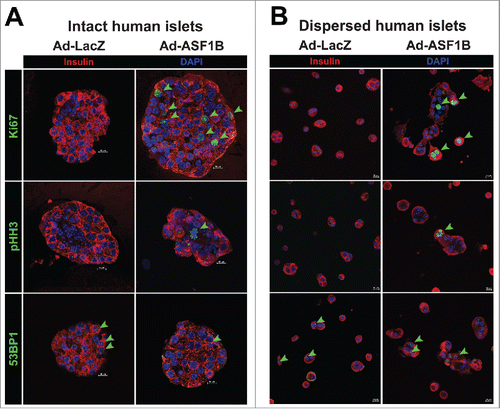
While our data strongly suggests that ASF1B induces β-cell proliferation, it does not conclusively rule out the possibility of other DNA-dependent processes (e.g., DNA damage repair pathways) that may have contributed to BrdU or [3H]-thymidine incorporation into islet DNA. To further validate our observations that ASF1B induces bona-fide mitotic progression, we evaluated human islets for expression of additional markers of proliferation, and a well-characterized marker of DNA damage. In ASF1B-transduced islets, several β-cells were positive for the nuclear antigen Ki67, which marks all phases of the cell cycle, and for pHH3 (S10), a phosphorylated core histone protein that selectively marks cells undergoing mitosis (). None of the β-cells in control islets were positive for either Ki67 or pHH3 (S10), consistent with their quiescent state. 53BP1-positive nuclear foci, an indicator of DNA damage response,Citation42 were not significantly different between islets treated with either Ad-LacZ or Ad-ASF1B (). Parallel studies were conducted on dispersed human islet preparations (), and yielded the same results as that observed for intact islets (). Similar observations were made using a different marker of DNA damage, γH2AX (Fig. S5C), providing further evidence that ASF1B promotes cellular proliferation and not DNA damage pathways, consistent with our FACS-based analysis of cell cycle progression illustrated in . Relative to Ad-LacZ, Ad-ASF1B caused a significant increase in the number of Ki67 and pHH3-positive nuclei (Fig. S5A), whereas the number of 53BP1 or γH2AX-positive foci were not different between Ad-LacZ and Ad-ASF1B (Fig. S5B). In summary, our data demonstrates that human β-cells expressing ASF1B undergo proliferation and exit mitosis successfully without experiencing any significant DNA damage.
Figure 4. ASF1B overexpression induces mitotic progression, but not DNA damage, in intact and dispersed human islets. Representative confocal images (60X) of (A) intact human islets (N = 20) and (B) dispersed human islet cells treated with Ad-LacZ or Ad-ASF1B, and labeled with the proliferation markers Ki67 and phospho-histone H3 (pHH3 S10) that mark cells through mitotic progression, and with the DNA damage marker, 53BP1 to mark nuclei undergoing DNA damage response (48 h). β-cells are identified by insulin (red), nuclei by DAPI (blue).
The histone binding capability of ASF1B is required to promote β-cell proliferation
We next investigated whether the histone-binding capacity of ASF1B is required for it to induce β-cell proliferation. A single amino acid substitution, valine-94 to arginine-94 (V94R), has been shown to abolish histone bindingCitation38 ().
Figure 5. Histone-binding mutant of ASF1B fails to promote β-cell proliferation. (A) Crystal structure of yeast ASF1-H3-H4 complex (PDB 2HUE); V94 of ASF1 is highlighted red. (B) Binding of HIRA and histone H3.3 was detected in anti-Twin-strep-tag (ASF1B) immunoprecipitates and total cell lysates from MIN6 cells transiently expressing GFP, WT-ASF1B or ASF1B-V94R mutant proteins. (C) Representative images (100X) of MIN6 β-cells expressing ASF1A or ASF1B (green) for WT (top panels) or the V94R mutants (bottom panels). Cells are co-stained for DAPI (nuclei, white), and insulin (red). (D) Flow cytometry analysis of cell cycle progression in MIN6 β-cells 48 h post-transfection with indicated plasmid-based constructs. Data represent means ± SEM, N = 3. *, GFP vs. ASF1B-WT and #, ASF1B-WT vs. ASF1B-V94R, P ≤ 0.05.
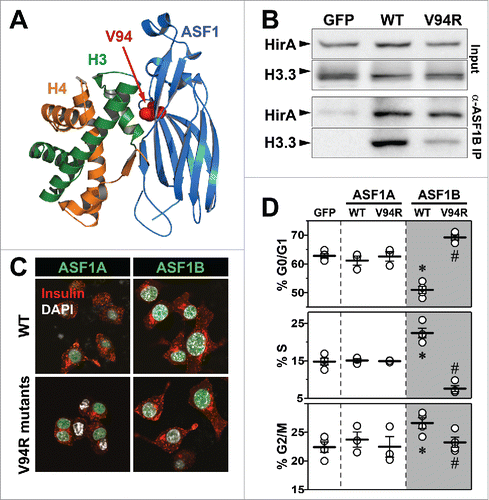
Like other post-mitotic non-proliferating cells, primary human β-cells also have little to no expression of ASF1B transcript or protein. However, the MIN6 β-cell line proliferates and expresses high levels of ASF1B, thus providing us with a good model system for testing the effect of the V94R mutation in ASF1A and ASF1B variants. Therefore, we expressed wild-type (WT) and the V94R mutant of ASF1A and ASF1B in MIN6 β-cells. WT, but not the V94R ASF1B bound endogenous histone H3.3, consistent with the non-histone binding mutation (). In contrast, both WT and V94R ASF1B bound equally well the endogenous secondary histone chaperone, HIRA. These results suggest that ASF1B contains distinct binding sites for histones and chaperones, in agreement with previously published reports.Citation41,43
Next, we asked whether loss of histone binding affected nuclear localization of ASF1 proteins. Transient overexpression of the WT and the V94R mutants for ASF1A and ASF1B yielded ∼100-fold increase in mRNA; protein levels were confirmed by western blot analysis (Fig. S6). Further, ASF1A and ASF1B proteins, irrespective of the V94R mutation, were localized almost exclusively to the nucleus, indicating that histone binding is not required for nuclear localization (). Despite similar cellular distributions, the ASF1 proteins differ significantly in their ability to promote cellular proliferation. We monitored cell proliferation via FACS-based DNA quantitation in cells transiently expressing WT or V94R versions of both ASF1 proteins. WT ASF1B, but not WT ASF1A, strongly promoted S-phase progression (). The V94R ASF1B mutant not only failed to induce proliferation but likely functioned as a dominant–negative for endogenous ASF1B, as it significantly reduced the population of cells in the S phase of the cell cycle (). Remarkably, ASF1A (both the WT and V94R mutant) did not have any significant effect on proliferation in MIN6 β-cells. These data indicate that despite ∼82% sequence homology, ASF1A and ASF1B are distinct in their regulation of cell cycle checkpoints in MIN6 β-cells, and that histone-binding is required for the effect of ASF1B on cellular proliferation.
The ASF1B-histone 3.3 complex is required for stimulating proliferation of human β-cells
Several studies have reported that the ASF1 proteins mediate RC and RI histone deposition using all 3 histone variants, H3.1, H3.2 and H3.3.Citation19,43,44 In humans, only H3.1 and H3.2 are utilized for RC deposition, leading to incorporation into nucleosomes during DNA synthesis.Citation43,45 In contrast, H3.3 is not utilized for RC deposition, yet is expressed in all phases of the cell cycle, including quiescent cells,Citation19,46,47 and is recruited for transcription-dependent gene regulation.Citation34
The WT version of ASF1B is capable of interacting with all H3 histone variants.Citation43,45 Therefore, we asked if one histone variant was preferentially utilized by ASF1B while stimulating β-cell proliferation in human islets. We transduced dispersed human islet cells with Ad-ASF1B, and Lentivirus (Lenti) encoding histones H3.1, H3.2, or H3.3A. Ad-GFP and control lentivirus were used as negative controls. In addition, we employed Lenti to overexpress a sh-RNA specific to human H3.3A to evaluate the loss-of-function for H3.3A on the ability of ASF1B to promote β-cell proliferation.
We first asked if modulating the histones yielded changes in secondary chaperone expression. Overexpression of all 3 histone H3 variants did not affect CAF1 expression levels (Fig. S7A). In contrast, the expression of HIRA mRNA was induced ∼75 and ∼150-fold by H3.2 and H3.3A, respectively (Fig. S7B). Surprisingly, suppression of endogenous H3.3A expression by sh-RNA yielded a significant induction in CAF1 expression (∼50-fold) and a small, but significant increase in HIRA expression (∼3-fold), suggesting that loss of H3.3A elicits a compensatory response by inducing CAF1 and HIRA expression.
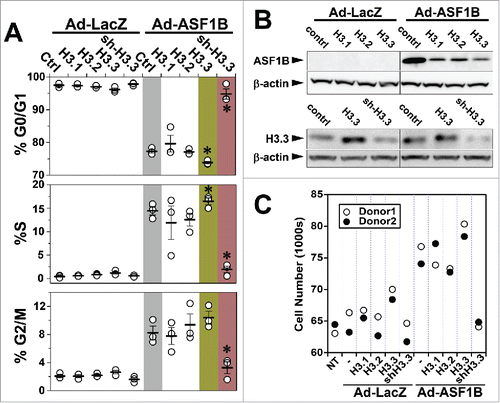
Overexpression of H3.1 and H3.2 alone, or in combination with ASF1B, did not alter the cell cycle profile of human β-cells (). In contrast, the stimulatory effect of ASF1B on S-phase progression was augmented when coupled with the overexpression of H3.3A. In control islets, ∼0.5% of the cells were in S-phase. In response to ASF1B, this proportion increased to ∼14%, and further increased to ∼16% when ASF1B was combined with H3.3A (). The population of cells in S-phase in ASF1B-expressing islets decreased significantly in response to H3.3 suppression; from ∼14% to ∼2%. A corresponding change in the population of cells in the G0/G1 and G2/M phases was also observed; ASF1B alone or in combination with H3.3A overexpression increased the number of cells in the G2/M phase, whereas H3.3 suppression increased accumulation of cells in the G0/G1 phase.
To extend our observations, we examined cell cycle progression by 2D FACS analysis using BrdU, as a fluorescent marker of DNA synthesis, and propidium iodide (PI) to monitor cellular DNA content. The 2D FACS data confirms that ASF1B promotes a significant increase in the proportion of cells in S-phase; from ∼1% to ∼11% for Ad-LacZ vs. Ad-ASF1B respectively. (Fig. S8A, B) In summary, our results demonstrate that ASF1B overexpression promotes S-phase progression in human β-cells.
To investigate whether modulated histone levels affect β-cell proliferation through ASF1B, we assessed the effect of histone overexpression and knock-down on ASF1B protein levels. Relative ASF1B protein abundance of islet cells overexpressing histone variants in combination with ASF1B was lower than that of ASF1B alone. However, ASF1B protein expression was not different between cells overexpressing H3.1, H3.2 or H3.3A in combination with ASF1B (). While all H3 variants have similar effect on ASF1B protein, the reduced ASF1B protein in single vs. double transduction could be an artifact of gross overexpression of 2 proteins simultaneously. In addition, the relative abundance of endogenous histone H3.3 was greatly reduced in response to sh-RNA H3.3A (). To determine if ASF1B-induced β-cell proliferation leads to a quantifiable increase in the number of β-cells, we developed an assay to determine cell number in response to Ad-LacZ vs. Ad-ASF1B. ASF1B lead to ∼1.5-fold increase in the number of cells. Further, the co-expression of histone H3.3A, augmented the effect of ASF1B, whereas suppression of endogenous H3.3A (via shRNA), abolished the effect of ASF1B (). In summary, our data indicates that ASF1B is a limiting factor in RI histone management that works synergistically with histone H3.3A to promote β-cell proliferation.
Discussion
Our study demonstrates that the histone chaperone ASF1B mediates H3.3-dependent transcriptional gene regulation, resulting in enhanced proliferation of human β-cells. ASF1 was first identified in yeastCitation48 where it was found to be essential for normal cell cycle progression.Citation31 We previously demonstrated that islet expression of Asf1b strongly correlated with the susceptibility of 2 mouse strains to obesity-induced diabetesCitation36 and with β-cell proliferation, consistent with studies in cancer showing that ASF1B plays a major role in cellular proliferation.Citation49
Figure 6. An interaction with H3.3 is necessary for ASF1B to induce human β-cell proliferation. (A) Flow cytometry analysis of cell cycle progression in disrupted human islets 48 h after indicated viral-mediated transductions. (B) Western blot illustrating relative protein abundance of ASF1B in islet cells expressing histone H3.1, H3.2 or H3.3 alone or in combination with Ad-LacZ (control) or ASF1B (upper panel); reduced protein abundance of histone H3.3 in response to sh-RNA mediated suppression (lower panel) N = 4 human donors. (C) Quantitation of change in cell number after ASF1B-mediated induction in human β-cell proliferation. Changes are measured using CyQuant assay and compared to control (no treatment, NT), N = 2 human donors.
Our RNA-sequencing of human islets and gene ontology analysis highlighted changes in gene regulation that supported a role for ASF1B in β-cell proliferation. ASF1B overexpression induced several genes involved in cell cycle regulation (see Table S3), including genes that mediate the G1/S transition, M phase, chromatin regulators, and cellular morphogenesis/organization. In addition, ASF1B repressed several genes involved in the regulation of apoptosis and programmed cell death. For example, SAL like 3 (SALL3) was induced ∼15-fold and is reported to be involved in cellular proliferation,Citation50 and interacts with DNMT3A to inhibit CpG island methylation in human cells. TRIAP1 (induced ∼4-fold) complexes with PRELI to prevent cellular apoptosis.Citation51 Gastrin-releasing peptide receptor (GRPR, induced ∼6-fold) has been linked with cell proliferationCitation52 and gastrin itself can stimulate β-cell survival and/or proliferation, perhaps through activation of the CCKB receptor.Citation53 The nuclear receptor NR1D1 was induced ∼2-fold upon ASF1B overexpression and is involved in circadian clock regulation, which suppresses the cell cycle inhibitory gene, p21WAF1/CIP1 in liver.Citation54 Interestingly, global (Clock) or pancreas-specific (Bmal1) disruption of key circadian regulators leads to β-cell dysfunction, including diminished β-cell proliferation.Citation55 It remains to be determined if ASF1B’s induction of NR1D1 links circadian regulation to improved β-cell proliferation by relieving the brake imposed by p21 protein levels in human islets. Overall, our transcriptomic analysis of human islets shows that a majority of the ASF1B-responsive genes fall in 2 distinct functional clusters; cellular proliferation and anti-apoptosis, suggesting that ASF1B regulates β-cell mass by a concerted stimulation of proliferation and suppression of cell death.
In addition to the genes discussed above, ASF1B overexpression caused a large change in the expression of several other genes. Myosin heavy chain gene (MYH8), a class II protein involved in regeneration and stem cell activation was induced ∼7-fold. Similarly, MLC1 (regulates leukoencephalopathy), POU4F1 (transcription factor associated with growth of cervical tumors) and NTS (regulates fat metabolism) were induced ∼5-fold. DNA binding protein NHLH2 that plays a role in cell differentiation was also induced ∼5-fold. Genes related to regulation of apoptosis and programmed cell death constituted a large cluster among the repressed genes. Other genes, such as heat shock protein HSPA6 and TP53TG3 (p53 target protein 3), were significantly repressed by ASF1B (∼4-fold and 3-fold respectively). CDON, a pro-apoptotic sonic hedgehog (shh) receptor and piggybac transposase PGBD4 were also downregulated by ∼2.5-fold. Expression of nuclear protein MDM1 which interacts with p53 was reduced by ∼2-fold in response to ASF1B overexpression. These gene expression changes may constitute downstream effects of ASF1B-induced β-cell proliferation.
The lack of a stimulatory effect of ASF1B on glucose-regulated insulin secretion or insulin content could be attributed to several factors. We believe that 48 - 72 hrs post-transduction is too short for newly formed β-cells to become fully functional β-cells. Similarly, Nkx6.1 has been shown to induce β-cell proliferation in human islets without positively effecting insulin secretion.Citation56 A recent review by Y. Liu et al. suggests that β-cells may not retain a differentiated phenotype necessary for regulated insulin secretion while replicating in-vitro.Citation57 Finally, the significant, but relatively small proportion of β-cells that are induced to replicate may not be sufficient to have an overall influence on insulin secretion when measured from the whole islet.
The ASF1B-evoked changes in the expression of cell cycle regulatory genes suggest that ASF1B promotes cellular proliferation. We demonstrated that ASF1B overexpression drives proliferation of human β-cells as judged by multiple measurements of islet cell proliferation. Interestingly, among the 2 paralogs of ASF1, ASF1A is expressed equally in replicating and quiescent cells, whereas ASF1B is expressed in proliferative tissues, but is essentially undetectable in quiescent cells.Citation58
A direct interaction between histones and ASF1 is required for histone transfer and nucleosome assembly during DNA replication and gene regulation.Citation59 ASF1A and ASF1B share a conserved N-terminal domain (residues 1-155), whereas their C-terminal regions are divergent.Citation60 The N-terminal domain includes the binding sites for histone H3-H4 dimer. Consequently, the histone-binding mutants of ASF1 (V94R),Citation38 as well as mutated histones that fail to bind ASF1,Citation43 lead to inefficient nucleosome assembly.
We examined the histone-binding requirements of ASF1B to promote β-cell proliferation. Whereas WT ASF1B was effective to promote β-cell proliferation, the V94R histone-binding mutant of ASF1B was unable to trigger proliferation, and functioned as dominant-negative for endogenous ASF1B function. While previous studiesCitation43,44 have suggested that ASF1 paralogs can share functional redundancy, the failure of ASF1A to influence β-cell proliferation highlights an interesting functional specialization between ASF1A and ASF1B in pancreatic β-cells. Our studies demonstrate that ASF1B, but not ASF1A, is capable of promoting β-cell proliferation, and that this capacity requires histone binding to ASF1B.
Ablation of ASF1 proteins, or loss of their histone-binding capacity, leads to mitotic and meiotic defects, and altered gene regulation.Citation43,49,61 Initially, ASF1 proteins were implicated in the formation of replication-coupling assembly factor (RCAF).Citation62 However, more recent reports show that ASF1B can directly interact with the B-domain of HIRA, suggesting that it may play a role in RI histone deposition.Citation41
Yeast ASF1 has been shown to bind all histone H3 variants.Citation19 H3.1 and H3.2 are expressed during the S phase of the cell cycle and deposited in a RC manner.Citation43 In contrast, H3.3 is ubiquitously expressed in all phases of the cell cycle and deposited in a RI manner.Citation47 Intriguingly, in HeLa cells, ASF1-HIRA interaction is not required for RI H3.3 deposition.Citation43 It is possible that other, yet to be identified factors require ASF1B and mediate RI deposition of H3.3 at target genes that lead to enhanced proliferation of human β-cells.
Replication-dependent and independent deposition of histone H3.1/H3.2 vs. H3.3 has been extensively studied,Citation19,35,43,63 but comparatively little is known about the chaperones that mediate this process. Our data suggest that in human β-cells, H3.3 is a major histone partner for ASF1B, and that overexpression of either protein is sufficient to stimulate β-cell proliferation. Interestingly, other H3 variants were ineffective in promoting proliferation, demonstrating a unique role for H3.3 in human β-cells. This may be explained in part by the fact that in terminally differentiated human β-cells, genes required for cell cycle activation are repressed. Therefore, the RC nucleosome assembly of constituent histones is a consequence rather than a cause of active proliferation.
In conclusion, we show that overexpression of ASF1B is sufficient to induce human β-cells to proliferate. While the capacity to bind histones is required for ASF1B, only histone H3.3 appears to be the required variant for its effect on cell cycle progression. This study is the first demonstration that a histone chaperone, along with a histone H3 variant, mediates transcriptional gene regulation and enhances proliferation of human β-cells.
Methods
Adenoviral construction
Human cDNA for ASF1B (Origene, SC113719, accession number NM_018154.2) was ligated into a shuttle plasmid and sequence-verified (Viraquest, VQ Ad5 CMV K-NpA). All viruses were generated and amplified by Viraquest (∼1010 pfu/ml).
Cell lines and cell culture
MIN6 pancreatic β-cells were cultured in DMEM tissue culture medium supplemented with 15% (v/v) fetal bovine serum (Sigma), 100 units/ml penicillin, and 0.1 mg/ml streptomycin at 37°C, 95/5% air/CO2.
Cell proliferation assay
In the CyQUANT assay 6×104 cells were seeded per well in triplicate in 96-well plates. Twenty-four hours later, cells were transduced with adenoviruses expressing LacZ, ASF1B or one of the histone variants as indicated. Forty-eight hours after infection, cells were harvested and the total DNA was quantified using the manufacturer’s instructions (CyQUANT, ThermoFisher Scientific,). Fluorescence (excitation 480 nm, emission 520 nm) was measured on a TECAN microplate reader. Similarly, a DNA standard curve was created by diluting lambda-DNA in 1 × CyQUANT buffer to give a range covering 1 to10 ng of DNA in 100μl of buffer. The standards were also processed and treated similarly to the test samples.
Flow cytometry
MIN6 β-cells or dispersed human islets were fixed with 100% ethanol and stained with propidium iodide (PtdIns) to measure cellular DNA content. Two-dimensional (2D) FACS assay was performed on dispersed human islets transuded with Ad-LacZ or Ad-ASF1B, alone or in combination with Lentiviruses expressing various histones. Cells were fixed with 100% ethanol, nuclei were isolated by pepsin digestion and treated with HCl. Nuclei were stained with FITC Anti-BrdU (1:200, 364103, Biolegend, CA) and propidium iodide (30 µg/µl, 81845, Sigma). Cells or nuclei were filtered through 40-μm sieves, and flow cytometry was performed on a 4-laser, LSRII (BD Biosciences). Data were analyzed with FlowJo and ModFit software.
[3H]-thymidine incorporation into DNA
48 hours after adenoviral infection, islets were incubated overnight in RPMI with 3H-thymidine (1 µCi/ml). Islets were then washed with ice-cold PBS. DNA and protein were precipitated by 10% trichloroacetic acid. The precipitate was solubilized in 0.3 N NaOH, and the radioactivity was measured using a liquid scintillation counter. A fraction of the solubilized protein was used to measure total protein by the Bradford assay. Sample radioactivity was normalized to protein content.
Human islet culture and dispersion
Islets from cadaveric human islets were obtained from the Integrated Islet Distribution Program (IIDP). A brief description of islet isolation procedure has been provided on IIDP website (https://iidp.coh.org/docs/CountingBrochure.pdf). Islets were cultured overnight in RPMI containing 8 mM glucose, 10% heat-inactivated FBS, and penicillin/streptomycin. For FACS analysis, islets were washed and digested with StemPro accutase cell dissociation reagent (Life Technologies, A11105-01) at 37°C for 15 minutes, filtered through 40-μm sieves, washed and cultured as indicated. See Table S1 for donor demographics.
Immunohistochemistry, immunocytochemistry and microscopy
Islets were washed in PBS, fixed in 10% formalin (3 hr, 4°C) followed by overnight incubation in PBS (4°C). Bio-Rad Affi-Gel Blue Gel beads (153-7301) were added to the islets to aid in their visualization during histological preparation. The mixture was fixed in 2% Agar/1% Formalin and then embedded in paraffin. Sections were dewaxed in xylene-EtOH and treated with an epitope retrieval solution (Vector Labs, H-3300). Sections were blocked (Dako, X0909) for 30 min, washed with PBS and probed with anti-insulin (1:500, I-8510, guinea pig, Sigma) and anti-BrdU (1:200, NA-61, Mouse, Calbiochem) primary antibodies. Secondary antibodies were Cy3-anti-guinea pig (1:500, 706-165-148, Jackson ImmunoResearch) and Alexafluor 488 anti-mouse (1:200, A11001, goat, Life Technologies). Staining was preserved and nuclei identified by DAPI (H-1200, Vector Labs). Images were acquired by EZ C1 software on Nikon A1R confocal microscope (Nikon Corp., Japan). In total 400 islets were evaluated for BrdU-positive cells/islet section. MIN6 β-cells were cultured on PDL-coated glass coverslips. Post-treatment, cells were washed with PBS and fixed in 4% PFA (30 minutes, RT). Coverslips were then washed with PBS and blocked in 10% donkey-serum and probed with anti-insulin (1:1000, I-8510, Sigma) and anti-Strep-Tag II (1:2000, 2-1507-001, IBA Life Sciences) antibodies. Slides were washed with PBS and stained with Cy3-anti-guinea pig (1:500, 706-165-148, Jackson ImmunoResearch and FITC-anti-mouse (1:200, 715-095-150, Jackson ImmunoResearch) antibodies. Nuclei were visualized by DAPI (H-1200, Vector Labs), and acquired as described above.
Immunoprecipitation and western blotting analysis
Cell lysates were prepared using cell lysis buffer (9803, CST, USA) containing protease inhibitor cocktail (469313200, Roche, Basel, Switzerland) following the manufacturer’s instructions. Immunoprecipitation and Western blotting analysis were carried out as described previously.Citation64
The following antibodies were used: Twin-Strep-Tag for Co-IP (2-1507-001, IBA life sciences, Goettingen, Germany); ASF1B for Western blotting (2902, CST); Histone H3.3 (09-838, EMD Millipore); HIRA (04-1488, EMD Millipore); and β-actin (3700S, Cell Signaling Technology, Danvers, MA, USA).
Isolation and quantitation of total RNA
RNA from dispersed human islets cells (transduced with Ad-LacZ, Ad-ASF1B or lentiviruses for histone H3.1, H3.2, H3.3 or sh-H3.3) was extracted using Qiagen RNeasy plus kit. Following extraction, RNA was used for cDNA synthesis (Applied Biosystems). The mRNA abundance was determined by quantitative PCR using FastStart SYBR Green (Roche Applied Science), and gene expression was calculated by comparative ΔCT method.
Lentiviral vectors
Sequence information for the lentiviruses expressing human histone H3.1, H3.2, H3.3A and sh-H3.3A/B are provided in Table S4.
Mouse models
The expression of Asf1a/b and Mki67 presented in was determined by Keller et al., 2008.Citation36 This study surveyed gene expression in islets of lean and genetically obese (Leptinob/ob) C57BL6 (B6) and BTBR mice at 4 or 10 weeks of age. Co-expression gene modules were identified in 6 different tissues, including pancreatic islets, across 3 contrast conditions; obesity, strain and age. A key module was identified in islets that was significantly enriched in cell cycle regulatory transcripts that predicted diabetes susceptibility in BTBR mice. Asf1b was included in this cell cycle module.
Plasmid transfection and viral transduction
MIN6 β-cell lines were transfected with Strep-tagged WT or V94R mutant variants of ASF1A and ASF1BCitation65 using Jetprime transfection reagent (Polyplus, Ilkrich, France) following manufacturer’s protocol.
Human islets were infected with adenovirus containing ASF1B or GFP at 200 multiplicity of infection (MOI, assuming 1000 cells/islet). Prior to adenoviral infection, islets were pre-treated in a Hanks Balanced Salt Solution without added calcium or magnesium, plus 2 mM EGTA for 3 minutes at 37°C to increase penetration and transduction efficiency. Islets were then treated with the adenovirus in 200 µl volume for 15 minutes, at 37°C, then cultured overnight in 3.5 ml RPMI without FBS. The next day, the islets were transferred to standard RPMI culture medium. Lentiviral transductions were performed on dispersed human islet cells in presence of 8 µg/ml polybrene (Hexadimethrine bromide – Sigma #H9268) for 12 hr. Forty-eight hours after transductions, cells were harvested for different experimental procedures.
RNA-sequencing and transcriptomic data analysis
RNA was isolated from 10 separate human islet samples (9 individual donors). The RNA was sequenced by indexed paired-end sequencing using an Illumina hi-seq 2500. Alignment was done using BowtieCitation66 and hg18 refseq annotations. Expression estimates were obtained from RSEMCitation67 with library size factors estimated using median normalization.Citation68 Differentially expressed (DE) genes and isoforms were identified using EBSeq.Citation69 Bowtie, RSEM and EBSeq (ver 1.3.2)Citation69 were ran using default parameters. Quality control on sequences was performed by FASTQC. Approximately 14.2 million aligned reads were obtained for each sample. Aligned reads were not filtered, however, “bad” reads were eliminated during alignment. To control the overall false discovery rate (FDR) at 5%, we defined a gene (isoform) as DE if its posterior probability exceeded 0.95. Enrichment tests were conducted using Allez.Citation70 All RNA-sequencing data has been deposited to NCBI GEO database (GSE86611).
Statistical analysis
Data are expressed as means ± SEM unless otherwise noted. Statistical significance (P < 0.05) was determined by paired or unpaired t-test as indicated (IGOR Pro version 6.3; WaveMetrics Inc., Lake Oswego, OR).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
P.K.P. and M.P.K. wrote the manuscript. P.K.P., M.P.K., M.E.R., C.-Y.W., D.S.S., N.L. performed experiments and analyzed the data. C.K. supervised statistical data analysis. P.W.L. provided key insights into the histone-binding characteristics of ASF1B. M.P.K. managed the project and all authors contributed to the design of the study. A.D.A. supervised the project and is the guarantor of the work, having had full access to all of the data and takes full responsibility for the integrity and accuracy of the analysis and interpretation of the data.
Supplementary files
Download Zip (17 MB)Acknowledgments
We thank Drs. Genevieve Almouzni and Zachary Gurard-Levin (Institut Curie, Paris) for comments on the manuscript, and for the generous gift of the WT and V94R mutant plasmids for ASF1A and ASF1B; Dr. Christopher Newgard (Duke University) for support and encouragement throughout the project.
Funding
This work was supported by the JDRF (3-PDF-2014-195-A-N) to P.K.P. and (17-2011-261) to A.D.A, and the NIH (DK066369, DK58037) to A.D.A. and (GM102756, UL1 RR025011) to C.K. Human islets were provided by the NIH-supported Integrated Islet Distribution Program.
ORCID
Pradyut K. Paul http://orcid.org/0000-0002-9922-4877
Christina Kendziorski http://orcid.org/0000-0002-0700-6267
Peter W. Lewis http://orcid.org/0000-0002-9816-7823
Mark P. Keller http://orcid.org/0000-0002-7405-5552
Related Research Data
References
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52:102-10; PMID:12502499; http://dx.doi.org/10.2337/diabetes.52.1.102
- Rankin MM, Kushner JA. Adaptive β-cell proliferation is severely restricted with advanced age. Diabetes 2009; 58:1365-72; PMID:19265026; http://dx.doi.org/10.2337/db08-1198
- Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y. Systemic regulation of the age-related decline of pancreatic β-cell replication. Diabetes 2013; 62:2843-8; PMID:23630298; http://dx.doi.org/10.2337/db13-0160
- Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic β cell fractional area and β cell turnover in human pregnancy. Diabetologia 2010; 53:2167-76; PMID:20523966; http://dx.doi.org/10.1007/s00125-010-1809-6
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal β cell mass. J Clin Invest 2004; 114:963-8; PMID:15467835; http://dx.doi.org/10.1172/JCI22098
- Parsons JA, Bartke A, Sorenson RL. Number and size of islets of Langerhans in pregnant, human growth hormone-expressing transgenic, and pituitary dwarf mice: effect of lactogenic hormones. Endocrinology 1995; 136:2013-21; PMID:7720649
- Xiao X, Chen Z, Shiota C, Prasadan K, Guo P, El-Gohary Y, Paredes J, Welsh C, Wiersch J, Gittes GK. No evidence for β cell neogenesis in murine adult pancreas. J Clin Invest 2013; 123:2207-17; PMID:23619362; http://dx.doi.org/10.1172/JCI66323
- Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocana A. Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes 2014; 63:819-31; PMID:24556859; http://dx.doi.org/10.2337/db13-1146
- Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 2012; 61:2205-13; PMID:22751699; http://dx.doi.org/10.2337/db12-0018
- Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocana A, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic β cell replication. Nat Med 2015; 21:383-8; PMID:25751815; http://dx.doi.org/10.1038/nm.3820
- Kassem S, Bhandari S, Rodriguez-Bada P, Motaghedi R, Heyman M, Garcia-Gimeno MA, Cobo-Vuilleumier N, Sanz P, Maclaren NK, Rahier J, et al. Large islets, β-cell proliferation, and a glucokinase mutation. N Engl J Med 2010; 362:1348-50; PMID:20375417; http://dx.doi.org/10.1056/NEJMc0909845
- Metukuri MR, Zhang P, Basantani MK, Chin C, Stamateris RE, Alonso LC, Takane KK, Gramignoli R, Strom SC, O'Doherty RM, et al. ChREBP mediates glucose-stimulated pancreatic β-cell proliferation. Diabetes 2012; 61:2004-15; PMID:22586588; http://dx.doi.org/10.2337/db11-0802
- Salpeter SJ, Klochendler A, Weinberg-Corem N, Porat S, Granot Z, Shapiro AM, Magnuson MA, Eden A, Grimsby J, Glaser B, et al. Glucose regulates cyclin D2 expression in quiescent and replicating pancreatic β-cells through glycolysis and calcium channels. Endocrinology 2011; 152:2589-98; PMID:21521747; http://dx.doi.org/10.1210/en.2010-1372
- Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature 2011; 478:349-55; PMID:21993628; http://dx.doi.org/10.1038/nature10502
- Cozar-Castellano I, Takane KK, Bottino R, Balamurugan AN, Stewart AF. Induction of β-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenovirus-mediated transfer of cyclin-dependent kinase-4 and cyclin D1. Diabetes 2004; 53:149-59; PMID:14693709; http://dx.doi.org/10.2337/diabetes.53.1.149
- Shu L, Sauter NS, Schulthess FT, Matveyenko AV, Oberholzer J, Maedler K. Transcription factor 7-like 2 regulates β-cell survival and function in human pancreatic islets. Diabetes 2008; 57:645-53; PMID:18071026; http://dx.doi.org/10.2337/db07-0847
- Fiaschi-Taesch N, Bigatel TA, Sicari B, Takane KK, Salim F, Velazquez-Garcia S, Harb G, Selk K, Cozar-Castellano I, Stewart AF. Survey of the human pancreatic β-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human β-cell replication and function in vivo. Diabetes 2009; 58:882-93; PMID:19136653; http://dx.doi.org/10.2337/db08-0631
- Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074-80; PMID:11498575; http://dx.doi.org/10.1126/science.1063127
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004; 116:51-61; PMID:14718166; http://dx.doi.org/10.1016/S0092-8674(03)01064-X
- Houlard M, Berlivet S, Probst AV, Quivy JP, Hery P, Almouzni G, Gerard M. CAF-1 is essential for heterochromatin organization in pluripotent embryonic cells. PLoS Genet 2006; 2:e181; PMID:17083276; http://dx.doi.org/10.1371/journal.pgen.0020181
- Le S, Davis C, Konopka JB, Sternglanz R. Two new S-phase-specific genes from Saccharomyces cerevisiae. Yeast 1997; 13:1029-42; PMID:9290207; http://dx.doi.org/10.1002/(SICI)1097-0061(19970915)13:11%3c1029::AID-YEA160%3e3.0.CO;2-1
- Adkins MW, Williams SK, Linger J, Tyler JK. Chromatin disassembly from the PHO5 promoter is essential for the recruitment of the general transcription machinery and coactivators. Mol Cell Biol 2007; 27:6372-82; PMID:17620413; http://dx.doi.org/10.1128/MCB.00981-07
- Akai Y, Adachi N, Hayashi Y, Eitoku M, Sano N, Natsume R, Kudo N, Tanokura M, Senda T, Horikoshi M. Structure of the histone chaperone CIA/ASF1-double bromodomain complex linking histone modifications and site-specific histone eviction. Proc Natl Acad Sci U S A 2010; 107:8153-8; PMID:20393127; http://dx.doi.org/10.1073/pnas.0912509107
- Chimura T, Kuzuhara T, Horikoshi M. Identification and characterization of CIA/ASF1 as an interactor of bromodomains associated with TFIID. Proc Natl Acad Sci U S A 2002; 99:9334-9; PMID:12093919; http://dx.doi.org/10.1073/pnas.142627899
- Schwabish MA, Struhl K. Asf1 mediates histone eviction and deposition during elongation by RNA polymerase II. Mol Cell 2006; 22:415-22; PMID:16678113; http://dx.doi.org/10.1016/j.molcel.2006.03.014
- Sutton A, Bucaria J, Osley MA, Sternglanz R. Yeast ASF1 protein is required for cell cycle regulation of histone gene transcription. Genetics 2001; 158:587-96; PMID:11404324
- Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell 2007; 27:393-405; PMID:17679090; http://dx.doi.org/10.1016/j.molcel.2007.07.011
- Grigsby IF, Rutledge EM, Morton CA, Finger FP. Functional redundancy of two C. elegans homologs of the histone chaperone Asf1 in germline DNA replication. Dev Biol 2009; 329:64-79; PMID:19233156; http://dx.doi.org/10.1016/j.ydbio.2009.02.015
- Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G. Regulation of replication fork progression through histone supply and demand. Science 2007; 318:1928-31; PMID:18096807; http://dx.doi.org/10.1126/science.1148992
- Sanematsu F, Takami Y, Barman HK, Fukagawa T, Ono T, Shibahara K, Nakayama T. Asf1 is required for viability and chromatin assembly during DNA replication in vertebrate cells. J Biol Chem 2006; 281:13817-27; PMID:16537536; http://dx.doi.org/10.1074/jbc.M511590200
- Tyler JK, Adams CR, Chen SR, Kobayashi R, Kamakaka RT, Kadonaga JT. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 1999; 402:555-60; PMID:10591219; http://dx.doi.org/10.1038/990147
- Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell 2005; 17:301-11; PMID:15664198; http://dx.doi.org/10.1016/j.molcel.2004.12.018
- Abascal F, Corpet A, Gurard-Levin ZA, Juan D, Ochsenbein F, Rico D, Valencia A, Almouzni G. Subfunctionalization via adaptive evolution influenced by genomic context: the case of histone chaperones ASF1a and ASF1b. Mol Biol Evolution 2013; 30:1853-66; PMID:23645555; http://dx.doi.org/10.1093/molbev/mst086
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 2002; 9:1191-200; PMID:12086617; http://dx.doi.org/10.1016/S1097-2765(02)00542-7
- Daniel Ricketts M, Frederick B, Hoff H, Tang Y, Schultz DC, Singh Rai T, Grazia Vizioli M, Adams PD, Marmorstein R. Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nature Communications 2015; 6:7711; PMID:26159857; http://dx.doi.org/10.1038/ncomms8711
- Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res 2008; 18:706-16; PMID:18347327; http://dx.doi.org/10.1101/gr.074914.107
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell 2007; 128:707-19; PMID:17320508; http://dx.doi.org/10.1016/j.cell.2007.01.015
- Mousson F, Lautrette A, Thuret JY, Agez M, Courbeyrette R, Amigues B, Becker E, Neumann JM, Guerois R, Mann C, et al. Structural basis for the interaction of Asf1 with histone H3 and its functional implications. Proc Natl Acad Sci U S A 2005; 102:5975-80; PMID:15840725; http://dx.doi.org/10.1073/pnas.0500149102
- Moshkin YM, Kan TW, Goodfellow H, Bezstarosti K, Maeda RK, Pilyugin M, Karch F, Bray SJ, Demmers JA, Verrijzer CP. Histone chaperones ASF1 and NAP1 differentially modulate removal of active histone marks by LID-RPD3 complexes during NOTCH silencing. Mol Cell 2009; 35:782-93; PMID:19782028; http://dx.doi.org/10.1016/j.molcel.2009.07.020
- Peng H, Nogueira ML, Vogel JL, Kristie TM. Transcriptional coactivator HCF-1 couples the histone chaperone Asf1b to HSV-1 DNA replication components. Proc Natl Acad Sci U S A 2010; 107:2461-6; PMID:20133788; http://dx.doi.org/10.1073/pnas.0911128107
- Tang Y, Poustovoitov MV, Zhao K, Garfinkel M, Canutescu A, Dunbrack R, Adams PD, Marmorstein R. Structure of a human ASF1a-HIRA complex and insights into specificity of histone chaperone complex assembly. Nat Struct Mol Biol 2006; 13:921-9; PMID:16980972; http://dx.doi.org/10.1038/nsmb1147
- Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 2013; 153:1266-80; PMID:23727112; http://dx.doi.org/10.1016/j.cell.2013.05.023
- Galvani A, Courbeyrette R, Agez M, Ochsenbein F, Mann C, Thuret JY. In vivo study of the nucleosome assembly functions of ASF1 histone chaperones in human cells. Mol Cell Biol 2008; 28:3672-85; PMID:18378699; http://dx.doi.org/10.1128/MCB.00510-07
- Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010; 140:678-91; PMID:20211137; http://dx.doi.org/10.1016/j.cell.2010.01.003
- Latreille D, Bluy L, Benkirane M, Kiernan RE. Identification of histone 3 variant 2 interacting factors. Nucleic Acids Res 2014; 42:3542-50; PMID:24393775; http://dx.doi.org/10.1093/nar/gkt1355
- Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 2010; 24:1253-65; PMID:20504901; http://dx.doi.org/10.1101/gad.566910
- Wu RS, Tsai S, Bonner WM. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell 1982; 31:367-74; PMID:7159927; http://dx.doi.org/10.1016/0092-8674(82)90130-1
- Munakata T, Adachi N, Yokoyama N, Kuzuhara T, Horikoshi M. A human homologue of yeast anti-silencing factor has histone chaperone activity. Genes Cells 2000; 5:221-33; PMID:10759893; http://dx.doi.org/10.1046/j.1365-2443.2000.00319.x
- Corpet A, De Koning L, Toedling J, Savignoni A, Berger F, Lemaitre C, O'Sullivan RJ, Karlseder J, Barillot E, Asselain B, et al. Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J 2011; 30:480-93; PMID:21179005; http://dx.doi.org/10.1038/emboj.2010.335
- Chai L. The role of HSAL (SALL) genes in proliferation and differentiation in normal hematopoiesis and leukemogenesis. Transfusion 2011; 51Suppl4:87S-93S; http://dx.doi.org/10.1111/j.1537-2995.2011.03371.x
- Potting C, Tatsuta T, Konig T, Haag M, Wai T, Aaltonen MJ, Langer T. TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab 2013; 18:287-95; PMID:23931759; http://dx.doi.org/10.1016/j.cmet.2013.07.008
- Cornelio DB, Roesler R, Schwartsmann G. Gastrin-releasing peptide receptor as a molecular target in experimental anticancer therapy. Ann Oncol 2007; 18:1457-66; PMID:17351255; http://dx.doi.org/10.1093/annonc/mdm058
- Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, Merlino GT, Schmidt EV, Brand SJ. Pancreatic gastrin stimulates islet differentiation of transforming growth factor α-induced ductular precursor cells. J Clin Invest 1993; 92:1349-56; PMID:8376589; http://dx.doi.org/10.1172/JCI116708
- Grechez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem 2008; 283:4535-42; PMID:18086663; http://dx.doi.org/10.1074/jbc.M705576200
- Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010; 466:627-31; PMID:20562852; http://dx.doi.org/10.1038/nature09253
- Schisler JC, Fueger PT, Babu DA, Hohmeier HE, Tessem JS, Lu D, Becker TC, Naziruddin B, Levy M, Mirmira RG, et al. Stimulation of human and rat islet β-cell proliferation with retention of function by the homeodomain transcription factor Nkx6.1. Mol Cell Biol 2008; 28:3465-76; PMID:18347054; http://dx.doi.org/10.1128/MCB.01791-07
- Liu Y, Mziaut H, Ivanova A, Solimena M. β-Cells at the crossroads: choosing between insulin granule production and proliferation. Diabetes Obesity Metab 2009; 11 Suppl 4:54-64; http://dx.doi.org/10.1111/j.1463-1326.2009.01107.x
- Polo SE, Theocharis SE, Klijanienko J, Savignoni A, Asselain B, Vielh P, Almouzni G. Chromatin assembly factor-1, a marker of clinical value to distinguish quiescent from proliferating cells. Cancer Res 2004; 64:2371-81; PMID:15059888; http://dx.doi.org/10.1158/0008-5472.CAN-03-2893
- Ishikawa K, Ohsumi T, Tada S, Natsume R, Kundu LR, Nozaki N, Senda T, Enomoto T, Horikoshi M, Seki M. Roles of histone chaperone CIA/Asf1 in nascent DNA elongation during nucleosome replication. Genes Cells 2011; 16:1050-62; PMID:21895891; http://dx.doi.org/10.1111/j.1365-2443.2011.01549.x
- Li Z, Gourguechon S, Wang CC. Tousled-like kinase in a microbial eukaryote regulates spindle assembly and S-phase progression by interacting with Aurora kinase and chromatin assembly factors. J Cell Sci 2007; 120:3883-94; PMID:17940067; http://dx.doi.org/10.1242/jcs.007955
- Messiaen S, Guiard J, Aigueperse C, Fliniaux I, Tourpin S, Barroca V, Allemand I, Fouchet P, Livera G, Vernet M. Loss of the histone chaperone ASF1B reduces female reproductive capacity in mice. Reproduction 2016; 151(5):477-89; PMID:26850882
- Mello JA, Sillje HH, Roche DM, Kirschner DB, Nigg EA, Almouzni G. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep 2002; 3:329-34; PMID:11897662; http://dx.doi.org/10.1093/embo-reports/kvf068
- Song Y, Seol JH, Yang JH, Kim HJ, Han JW, Youn HD, Cho EJ. Dissecting the roles of the histone chaperones reveals the evolutionary conserved mechanism of transcription-coupled deposition of H3.3. Nucleic Acids Res 2013; 41:5199-209; PMID:23563152; http://dx.doi.org/10.1093/nar/gkt220
- Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, Kumar A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol 2010; 191:1395-411; PMID:21187332; http://dx.doi.org/10.1083/jcb.201006098
- Duro E, Lundin C, Ask K, Sanchez-Pulido L, MacArtney TJ, Toth R, Ponting CP, Groth A, Helleday T, Rouse J. Identification of the MMS22L-TONSL complex that promotes homologous recombination. Mol Cell 2010; 40:632-44; PMID:21055984; http://dx.doi.org/10.1016/j.molcel.2010.10.023
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/10.1186/gb-2009-10-3-r25
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011; 12:323; PMID:21816040; http://dx.doi.org/10.1186/1471-2105-12-323
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106; PMID:20979621; http://dx.doi.org/10.1186/gb-2010-11-10-r106
- Leng N, Li Y, McIntosh BE, Nguyen BK, Duffin B, Tian S, Thomson JA, Dewey CN, Stewart R, Kendziorski C. EBSeq-HMM: a Bayesian approach for identifying gene-expression changes in ordered RNA-seq experiments. Bioinformatics 2015; 31(16):2614-22; PMID:25847007
- Newton MA, He Q, Kendziorski C. A model-based analysis to infer the functional content of a gene list. Stat Appl Genet Mol Biol 2012; 11(2); PMID:22499692