
ABSTRACT
Homologous recombination (HR) is a conserved process that maintains genome stability and cell survival by repairing DNA double-strand breaks (DSBs). The RAD51-related family of proteins is involved in repair of DSBs; consequently, deregulation of RAD51 causes chromosomal rearrangements and stimulates tumorigenesis. RAD51C has been identified as a potential tumor suppressor and a breast and ovarian cancer susceptibility gene. Recent studies have also implicated estrogen as a DNA-damaging agent that causes DSBs. We found that in ERα-positive breast cancer cells, estrogen transcriptionally regulates RAD51C expression in ERα-dependent mechanism. Moreover, estrogen induces RAD51C assembly into nuclear foci at DSBs, which is a precursor to RAD51 complex recruitment to the nucleus. Additionally, disruption of ERα signaling by either anti-estrogens or siRNA prevented estrogen induced upregulation of RAD51C. We have also found an association of a worse clinical outcome between RAD51C expression and ERα status of tumors. These findings provide insight into the mechanism of genomic instability in ERα-positive breast cancer and suggest that individuals with mutations in RAD51C that are exposed to estrogen would be more susceptible to accumulation of DNA damage, leading to cancer progression.
Introduction
Homologous recombination (HR) is a conserved process that maintains genome stability and cell survival by repairing DNA double-strand breaks (DSBs). DSBs arise from V(D)J recombination, meiosis, class switch recombination, exposure to reactive oxygen species, ionizing radiation (IR), ultraviolet light or chemotherapeutic agents as well as DNA replication. Unrepaired DSBs represent the most detrimental form of DNA damage and lead to accumulation of chromosome aberrations that may lead to immunodeficiency, neurodegeneration and cancer susceptibility. The importance of proper HR to DSB repair is further supported by findings that impaired HR is associated with genetic diseases such as ataxia telangiectasia, Nijmegen breakage syndrome, Fanconi anemia (FA) and Bloom's syndrome.Citation1,2 Importantly, mutations in HR-related genes are associated with tumorigenesis and cause accumulation of unrepaired DSBs leading to increased genomic instability and cancer. Breast cancer is the most common cancer affecting women, and ∼5% of cases are due to inherited mutations in BRCA1 and BRCA2 genes, encoding for tumor suppressors that are involved in HR and DSB repair. Heterozygous mutation of BRCA1 or BRCA2 increases the risk of developing breast cancer by 80% and ovarian cancer by 40%.Citation3-6
RAD51 is a small monomeric protein that assembles into long helical polymers on single strand DNA at the break site during HR and is important for DSB repair.Citation7 Deregulation of RAD51 in human cells causes chromosome rearrangements and stimulates tumorigenesis.Citation8 RAD51 paralogs is a family of proteins that help RAD51 recruitment to the DNA break sites and include RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3.Citation9-13 These paralogs share 20–30% of identity at the amino acid level and can assemble into 2 distinct complexes: RAD51B/RAD51C/RAD51D/XRCC2 complex and RAD51C/XRCC3 complex, with RAD51C being the only member that is part of both complexes.Citation8,14,15 Mutation of Rad51 paralogs in mice causes early embryonic lethality and accumulation of unrepaired DNA damage,Citation16,17 highlighting their function in preserving genomic integrity. Abrogation of their function in Chinese hamster ovary (CHO) cells or chicken DT40 B-lymphocytes sensitizes cells to ionizing radiation (IR) and DNA damaging agents such as mitomycin C, cisplatin and camptothecin.Citation18-26 Additionally, RAD51-deficient cells exhibit increased chromosomal aberrations, abnormal centromere number, reduced frequency of DSB repair and reduced sister chromatid exchanges. Knockdown of RAD51 expression was able to inhibit cancer cell migration as well as tumor growth and metastasis,Citation27 highlighting the role of RAD51 in triple negative breast cancers (TNBC). Specifically, in TNBC, it has been shown that co-inhibition of RAD51 together with p38 reduced cell proliferation and may be a novel clinical strategy.Citation28
RAD51C, which has a role in HR and RAD51 recruitment to DSBs, has been recently identified as a potential tumor suppressorCitation29 and a breast and ovarian cancer susceptibility gene.Citation30-32 RAD51C appears to have a uniquely important role in breast cancer. RAD51C is localized to the chromosomal region 17q23, an amplicon present in high copy number in breast cancer.Citation33-35 An additional link between RAD51C and breast cancer stems from direct interaction of BRCA2 with RAD51 complex, which directs BRCA2 to the sites of DNA damage.Citation36,37 BRCA1 localizes with RAD51 into nuclei foci,Citation38 further implicating RAD51 paralogs in both DSB repair and breast cancer.
Upon DNA damage caused by IR, RAD51C accumulates into nuclear foci and co-localizes with RAD51. Moreover, RAD51C is thought to regulate the translocation of RAD51 from the cytoplasm to the nucleus via the nuclear localization signal.Citation39,40 Rad51c deletion in mouse embryonic fibroblasts (MEFs) also results in reduction in sister chromatid exchanges and failure to proliferate, indicating that RAD51C is required for inter-sister chromatid recombination repair in mice.Citation41 Furthermore, RAD51C-mutated breast and ovarian cancers have similar histopathology to that of BRCA2-defective cancers indicating the involvement of RAD51C in recombinational repair.Citation30 Additionally, recent clinical studies linked germline mutation in RAD51C with development of FA-like disorder,Citation42 further underscoring the crucial role of RAD51C in DSB repair.
In addition to its role in DSB repair, RAD51C also acts as a mediator of checkpoint signaling. In response to DNA damage, RAD51C facilitates phosphorylation of checkpoint kinase 2 (CHK2) by ataxia-telangiectasia mutated (ATM) and delays cell cycle progression.Citation43 CHK2 is a cancer susceptibility gene mutations in which have been identified in familial breast and prostate cancersCitation44-47 that relays DNA damage signaling and regulates cell cycle progression, DNA repair and progression to senescence or apoptosis.Citation48 Additionally, in HR-mediated DSB repair, CHK2 phosphorylates BRCA1Citation49 further connecting the proper regulation of DSB repair and checkpoint signaling in cancer.
Recent studies implicated estrogen as a DNA-damaging agent that causes DSBs. It was found that treatment of breast cancer cells with estrogen induced DSBs and caused co-localization with RAD51,Citation50 suggesting that defects in DSB repair could contribute to ERα-positive breast cancer pathogenesis. RAD51C is localized at the chromosomal region 17q23, which appears to be a hotspot for estrogen-driven gene expression.Citation51 Importantly, expression of several genes within this amplicon have been shown to be regulated by estrogen, including RPS6KB1, PPM1D and MIR21 - all of which have oncogenic roles in breast cancer.Citation52-54 Based on these findings, we hypothesized that estrogen may regulate RAD51C expression and DSB repair. Indeed, we found that in ERα-positive breast cancer cells, estrogen transcriptionally regulates RAD51C expression in ERα-dependent mechanism. Moreover, estrogen induces RAD51C assembly into nuclear foci at DSBs, which is a precursor to RAD51 recruitment to the nucleus. Finally, we found a prognostic correlation between ERα-positive breast cancer and RAD51C expression, further establishing the clinical significance of these findings.
Results
Estrogen induces RAD51C expression in ERα-positive but not ERα-negative breast cancer cells
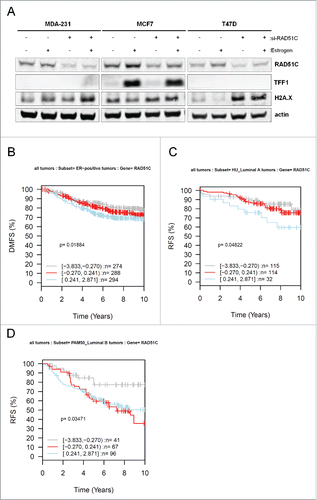
To confirm that estrogen induces DNA damage, ERα-positive and ERα-negative breast cancer cells were treated with estrogen for 30 minutes, which was sufficient to induce DNA damage response signaling as analyzed by phosphorylation of ATM on S1981 and Chk2 on T68 (). Although phosphorylation of ATM on S1981 was preferentially induced in ERα-positive breast cancer cells, phosphorylation of Chk2 on T68 was activated irrespective of the ERα status of the cell, indicating that this process does not require estrogen receptor signaling. Estrogen effect was specific to activation of DNA-damage response pathway and not general cytoplasmic kinase signaling as levels of phospho-ERK remained unchanged. Previous work has shown that estrogen induces localization of RAD51 to the sites of DNA damage, and RAD51C regulates nuclear translocation of RAD51.Citation39,40 Because RAD51C is localized to the 17q23 region, which contains estrogen regulated genes, we wanted to test whether estrogen can induce RAD51C expression in ERα-dependent manner. As shown in and quantified in , estrogen treatment caused upregulation of RAD51C expression in the ERα-positive MCF7, T47D, and ZR-75-1 breast cancer cells. As expected, expression of TFF1, a known estrogenically-regulated gene is induced solely in ERα-positive MCF7 and ZR 75-1 cells and not ERα-negative cells (). T47D cells express TFF1 at much lower levels reflecting heterogeneity among ER-positive cell lines. Most importantly, the effect of estrogen on RAD51C expression appeared dependent on the presence ERα, as in ERα-negative MDA-MB-231, −436, and −468 cells, RAD51C levels were not affected by estrogen treatment ().
Figure 1. ERα regulates RAD51C expression in an estrogen dependent manner. (A) MDA-MB-231, −468, −436, MCF7, T47D and ZR 75-1 cells were grown in phenol red-free media with 10% charcoal-stripped FBS for 3 d and either serum starved or treated with estrogen for 30 min as indicated. Lysates were generatedCitation67 and the indicated proteins were detected by immunoblot. (B) MDA-MB-231, −468, −436, MCF7, T47D and ZR 75-1 cells were grown in phenol red-free media with 10% charcoal-stripped FBS for 3 d and either serum starved or treated with 10nM estrogen for 24 hr as indicated. Lysates were generated as described in “Materials and Methods” and the indicated proteins were detected by immunoblot. (C) Quantification of RAD51C protein levels normalized to actin from (B) was performed using Odyssey Image Studio Version 4.0 and graphed using Excel.
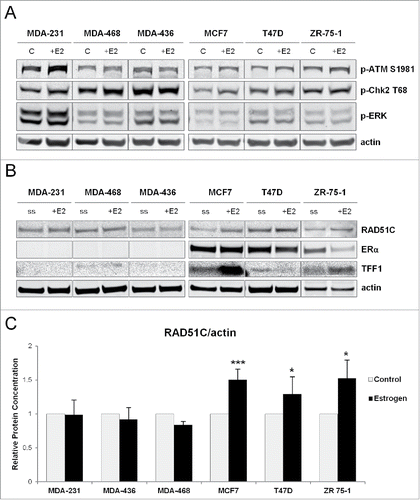
Estrogen induces RAD51C focus formation in ERα-dependent manner
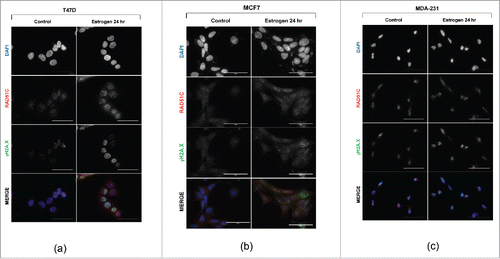
Since estrogen has previously been shown to induce DNA double-strand breaks, and we observed that estrogen regulates RAD51C protein expression, we hypothesized that estrogen can also induce nuclear RAD51C focus formation in ERα-dependent manner. Using immunofluorescence, we observed that estrogen induces γH2A.X foci in the nucleus of T47D breast cancer cells, a marker of DNA double-strand breaks, as previously shown.Citation50 Importantly, similar to estrogen's induction of γH2A.X foci formation, RAD51C foci were also induced by estrogen treatment (). This finding was further observed in another ERα-positive breast cancer cell line, MCF7 (). To test for the requirement of ERα in estrogen-induced RAD51C focus formation, MDA-MB-231 cells, an ERα-negative breast cancer cell line was used. As expected, MDA-MB-231 cells did not show induction of γH2A.X or RAD51C foci upon estrogen treatment (), indicating that RAD51C localization to the DNA double-strand breaks requires ERα.
Figure 2. Estrogen induces RAD51C foci assembly. (A) T47D cells were serum-starved or stimulated with estrogen for 24 hr as indicated. Immunofluorescence was performed as described in “Materials and Methods.” Scale bar represents 50 μm. (B) MCF7 cells were treated and processed as described in (A). (C) MDA-MB-231 cells were treated and processed as described in (A).
Estrogen directly regulates RAD51C expression via ERα
To confirm that the regulation of RAD51C expression is mediated by ERα, we downregulated ERα expression using a combination of 2 siRNA against ERα (Supplementary ) and treated the cells with estrogen for 24 hr. RT-qPCR analysis of MCF7 cells showed upregulation of mRNA for RAD51C () and TFF1 () in the presence of estrogen, while cells transfected with siRNA against ERα showed reduced upregulation of RAD51C when stimulated with estrogen. This effect on RAD51C was further observed on the level of protein in MCF7, ZR-75-1 cells and T47D cells, indicating that RAD51C expression is regulated by estrogen resulting in increased protein product, while ERα knockdown reduced RAD51C protein induction ().
Figure 3. ERα transcriptionally regulates RAD51C expression in estrogen-dependent manner. (A) MCF7 cells were transfected with scambled siRNA or siRNA against ERα and treated with estrogen for 48 hr, as indicated. RT-qPCR was performed as described in “Materials and Methods” and data was plotted using Excel. **p < 0.001; NS, non-statistically significant; n = 3. (B) MCF7 cells were treated as described in (A), RT-qPCR was performed as described in “Materials and Methods” and data was plotted using Excel. **p < 0.001; NS, non-statistically significant; n = 3. (C) MDA-MB-231, −468, −436, MCF7, T47D and ZR 75-1 cells were serum starved or treated with estrogen for 24 hr, as indicated. Lysates were generated as described in “Materials and Methods” and the indicated proteins were detected by immunblot. (D) MCF7 cells were transfected, stimulated with estrogen for 24 hr as indicated and Luciferase reporter assay was performed as described in “Materials and Methods.” Data was plotted using Excel. **p < 0.001. (E) MCF7 cells were transfected, stimulated with estrogen for 24 hr as indicated and Luciferase reporter assay was performed as described in “Materials and Methods.” Data was plotted using Excel. **p < 0.001; n = 3. (F) MDA-MB-231 and MCF7 cells were either serum starved, treated with estrogen for 24 hr or pre-treated with estrogen for 30 min following treatment with tamoxifen for 24 hr. Lysates were generated as described in “Materials and Methods” and indicated proteins were detected by immunoblot. (G) MDA-MB-231 and MCF7 cells were either serum starved, treated with estrogen for 24 hr or pre-treated with estrogen for 30 min following treatment with fulvestrant for 24 hr. Lysates were generated as described in “Materials and Methods” and indicated proteins were detected by immunoblot.
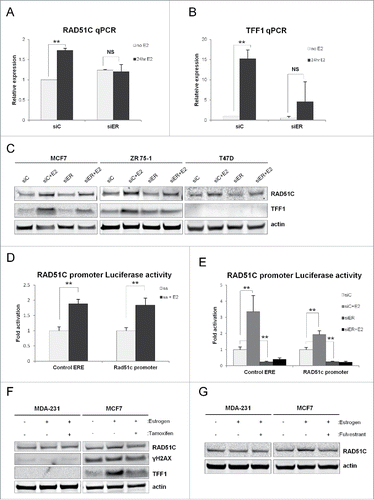
To confirm that estrogen regulates RAD51C on the transcriptional level, MCF7 cells were transfected with a construct whereby expression of renilla luciferase is driven by the proximal RAD51C promoter. A construct encoding for firefly luciferase under the control of 3 estrogen response elements (EREs) was used as an internal control for ER-mediated transcription. As shown in , estrogen stimulation significantly increased the expression of luciferase controlled by the RAD51C promoter. Additionally we observed that downregulation of ERα expression using siRNA significantly reduced expression of luciferase controlled by the RAD51C promoter and the addition of estrogen was not able to counteract this effect (), indicating that estrogen transcriptionally regulates RAD51C via ERα.
To further confirm the role of ERα mediated regulation of RAD51C, MCF7 and MDA-MB-231 cells were treated with estrogen in conjunction with either tamoxifen, a selective estrogen receptor modulator, or fulvestrant, a selective estrogen receptor degrader, to disrupt the action of estrogen. Treatment of MCF7 cells with either tamoxifen () or fulvestrant () was able to block estrogen mediated upregulation of RAD51C. Furthermore, this effect was dependent on ERα, as RAD51C levels remained unchanged in MDA-MB-231 cells. Induction of TFF1 expression served as control.
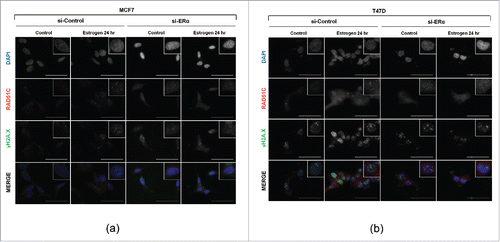
To validate that ERα is necessary for estrogen induced RAD51C focus formation, the expression of ERα was downregulated in MCF7 cells using siRNA, and cells were stimulated with estrogen for 24 hr. Compared to cells transfected with scrambled siRNA that showed RAD51C and γH2A.X focus formation upon treatment with estrogen, MCF7 cell transfected with siRNA against ERα did not exhibit formation of either RAD51C or γH2A.X foci upon estrogen treatment (). This finding was also confirmed in another ERα-positive cell line, T47D ().
Figure 4. Estrogen induces RAD51C foci assembly in ERα dependent manner. (A) T47D cells were transfected with scrambled siRNA, or siRNAs against ERα and either serum-starved or stimulated with estrogen for 24 hr. Immunofluorescence was performed as described in “Materials and Methods.” Scale bar represents 50 μm. (B) MCF7 cells were treated and processed as described in (A).
RAD51C expression is prognostic of poor clinical outcome in ERα-positive breast cancer
Owing to the fact that RAD51C is a breast cancer susceptibility gene, we wanted to investigate the effect of estrogen in cells transfected with siRNA against RAD51C (Supplementary ). As expected, estrogen treatment upregulated RAD51C levels in ERα-positive and not ERα-negative breast cancer cells () and this effect was diminished in cells transfected with siRNA against RAD51C. Interestingly, γH2A.X levels were elevated in cells with reduced levels of RAD51C, indicating that these cells experience DNA damage. To explore the clinical significance between RAD51C expression and ERα status of breast tumors, we used the Gene expression-based Outcome for Breast cancer Online (GOBO) database to examine the association between the RAD51C expression in ERα-positive breast tumors and patient outcome. The GSA-Tumor analysis of RAD51C expression revealed a worse clinical outcome in ER-positive tumors with regard to Distant Metastasis Free Survival (DMSF) endpoint, as shown in . The poor prognostic value of RAD51C was further confirmed using Relapse-Free Survival (RFS) endpoint when tumors where stratified based on the luminal A and B (ERα-positive like) breast cancer molecular subtypeCitation55 ().
Figure 5. GSA-Tumor analysis of RAD51C shows worse clinical prognosis for ERα-positive tumors. (A) MDA-MB-231, MCF7 and T47D cells were transfected with scrambled siRNA or siRNA against RAD51C and treated with estrogen for 48 hr, as indicated. Lysates were generated as described in “Materials and Methods” and the indicated proteins were detected by immunblot. (B) Kaplan-Meier analysis using DMFS of ERα-positive tumors separated into 3 categories with respect to RAD51C expression. (B) Kaplan-Meier analysis using RFS of Luminal A tumors separated into 3 categories with respect to RAD51C expression. (C) Kaplan-Meier analysis using RFS of Luminal A tumors separated into 3 categories with respect to RAD51C expression.
Discussion
Breast cancer is a heterogeneous disease with involvement of different cellular mechanisms and signaling pathways.Citation56 Identification of the molecular targets that are altered is a crucial challenge for our understanding of breast cancer pathogenesis and facilitating the design of tailored treatments that are effective at stopping breast cancer progression and preventing cancer recurrence. ERα status of breast cancer is an important marker for selecting patients for treatment with endocrine therapy such as selective estrogen receptor modulators (e.g- tamoxifen), selective estrogen receptor degraders (e.g. fulvestrant) or aromatase inhibitors (such as letrozole). However, only 35–70% of patients respond to endocrine therapy and resistance develops in most cases, in part due to activation of growth factor signaling pathways.Citation57 Another pathway that is often attenuated in breast cancer is the DSB repair with main focus on germline loss of function of tumor suppressors BRCA1 and BRCA2 leading to hereditary breast and ovarian cancer progression. Mutations in DSB repair genes lead to accumulation of DSBs, genome instability and tumorigenesis.
RAD51C is a recently identified high penetrance cancer susceptibility gene,Citation30-32 and germline mutations in RAD51C predispose to breast and ovarian cancer with high similarity to patients carrying BRCA1 and BRCA2 mutations. In the current work we identified a novel mechanism of estrogen dependent DSB repair, whereby estrogen regulates RAD51C via ERα on a transcriptional level, demonstrating a direct link between ERα signaling and HR pathways. A recent study describing Rad51c- and Trp53-double-mutant mice showed that these mice have an accelerated production of mammary carcinomas.Citation58 Similar to our results, these tumors exhibited high levels of genomic instability and DNA damage and most interestingly, the tumors retained expression of ERα, which is similar to the luminal-like phenotype described in RAD51C mutant breast cancers.Citation59 Therefore, the estrogen-dependent DNA damage and repair mechanism we described here could be responsible for the phenotype observed in vivo. Paradoxically, estrogen appears to induce DNA damage, either by direct oxidative damage or indirectly, by promoting progression into S-phase, while simultaneously activating the expression of DNA damage repair genes, such as RAD51C, which may be a homeostatic mechanism that prevents accumulation of estrogen-induced DNA damage. This finding adds a potential new link to the relationship between lifetime estrogen exposure and breast malignancy. Clinically, these findings may be extremely significant because ERα-positive and estrogen dependent breast cancer tumors would incur estrogen-induced DNA damage, thus increased expression of RAD51C could be indicative of increased genomic instability and worse disease outcome. Moreover, individuals with inactivating mutations in RAD51C would be more susceptible to accumulation of unrepaired estrogen-induced DNA damage, increased genomic instability and cancer progression, similar to deficiencies in BRCA1/2 and RAD51.Citation60-62 Estrogen is sometimes used clinically for treatment of breast cancer because of its apoptosis-inducing capacity.Citation63 It would be interesting to determine whether in this setting, high-dose estrogen may contribute to synthetic lethality in combination with DNA damaging agents such as cisplatin and PARP inhibitors.Citation64,65 Thus RAD51C may serve as a prognostic marker for disease severity in ER-positive breast cancer, as well as a marker for sensitivity to DNA damaging chemotherapeutics.
Materials and methods
Cell culture and treatment
MDA-MB-231, MDA-MB-436, MDA-MB-468, MCF7, ZR 75-1 and T47D cells were cultured in a humidified incubator with 5% CO2 at 37°C in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). For experiments, cells were grown in phenol red-free media with 10% charcoal-stripped FBS for 3 d. Where indicated, cells were starved in serum-free media for 24 hr and stimulated with 10nM estrogen (in ethanol), 100 nM 4-hydroxy-tamoxifen (in ethanol), or 100 nM fulvestrant (in DMSO).
siRNA transfection: siRNAs against ERα (HSC.RNAI.N000125.12.5 and HSC.RNAI.N000125.12.6), RAD51C (HSC.RNAI.N058216.12.1 and HSC.RNAI.N058216.12.2) and scrambled controls were ordered from Integrated DNA technologies. 25 pmol of siRNA was transfected using Lipofectamine® RNAiMAX Transfection Reagent (Invitrogen) according to manufacturer's protocol. Cells were lysed 48 h post-transfection.
Constructs: Firefly luciferase-3ERE reporter plasmid was previously described.Citation66 Renilla luciferease-RAD51C promoter was purchased from SwitchGear Genomics.
Immunofluorescence
Cells were plated on 18mm Poly-L-Lysine coated cover slips (Fisher Scientific). Following treatment, cells were fixed in 4% parafalmaldehyde and permeabilized in 0.2% Triton-X. After blocking and incubation with primary (RAD51C from Abcam, ab72063 and ERα from Santa Cruz Biotechnology, sc-8005) and secondary antibodies (Alexa Fluor 55 donkey anti-rabbit IgG, A31572 and Alexa Fluor 488 donkey anti-mouse IgG, A21202; all from Invitrogen), coverslips were mounted using DAPI-Fluoromount G mounting media (Southern Biotech, 0100-20). Images were collected using EVOS FL Auto microscope (Invitrogen) under 60X magnification.
Cell lysis
Cells were lysed in ice-cold buffer containing 10 mM KPO4, 1 mM EDTA, 10 mM MgCl2, 50 mM β-glycerophosphate, 5 mM EGTA, 0.5% Nonidet P-40 [NP-40], 0.1% Brij 35, 1 mM sodium orthovanadate, 40 µg/ml phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 5 µg/ml pepstatin, 10 µg/ml aprotinin. Lysates were cleared of insoluble material by centrifugation at 15,000g for 10 min at 4°C. Protein concentration in cell extracts was measured by Bradford reagent (BioRAD) according to the manufacturer's protocol using Eppendorf BioPhotometer (Eppendorf). Samples were equalized for protein concentration and denatured using LDS Sample buffer and Reducing agent (Invitrogen) at 70°C for 10 min. Samples were resolved using Bis-Tris Plus gels (Invitrogen) and transferred onto nitrocellulose membrane (GE Healthcare). Membranes were probed with the following primary antibodies: RAD51C (sc56214 Santa Cruz Biotechnology), p-ATM S1981 (5883 Cell Signaling Technologies), p-Chk2 T 68 (2197 Cell Signaling Technologies) p-ERK (4695 Cell Signaling), γH2A.X (05–636 Millipore), ERα (sc543 Santa Cruz Biotechnology), actin (sc1615 Santa Cruz Biotechnology), and TFF1 (12419S Cell Signaling Technologies).
Signal detection and quantification was accomplished using IRDye-conjugated anti-rabbit (LI-COR, 827-08365), anti-mouse (LI-COR, 926-68070) or anti-goat (LI-COR, 926-68074) secondary antibodies using Odyssey infrared detection instrument (LI-COR). All immunoblots were performed at least thrice to ensure reproducibility.
Luciferase reporter assay
MCF7 cells were transiently transfected with a Rad51c-promoter Renilla luciferase construct (SwitchGear Genomics) and a reporter plasmid containing 3 estrogen response elements (ERE) controlling expression of firefly luciferase, as previously described.Citation66 siRNA against ERα was co-transfected, as indicated in figure legends. Luciferase expression following cell treatment with estrogen was assayed using a dual luciferase kit and GloMax® 20/20 Luminometer (Promega). Assays were performed in triplicates and results were analyzed and plotted using Excel.
Quantitative RT-PCR
RNA was isolated using RNeasy® Mini Kit (Qiagen) and 1 µg of RNA was reverse transcribed into cDNA using iScript™ cDNA Synthesis Kit (BioRAD) and C1000 thermal cycler (BioRAD). For qPCR, cDNA was amplified with iQ™ SYBR® Green Supermix (BioRAD) in CFX96™Real-Time PCR Detection System (BioRAD, Hercules, CA) with CFX Manager analysis on-board software. TFF1 F: 5′ ATC GAC GTC CCT CCA GAA GAG 3′; TFF1 R: 5′ CTC TGG GAC TAA TCA CCG TGC TG 3′; 18s F: 5′ TTC GAA CGT CTG CCC TAT CAA 3′; 18s R: 5′ ATG GTA GGC ACG GCG ACT A 3′; Rad51c F: 5′ GGA TTT GGT GAG TTT CCC GC 3′; Rad51c R: 5′ TCT TTG CTA AGC TCG GAG GG 3′
Statistical analysis
Data are presented as mean ± S .D. and n = 3. Statistical significance was determined by paired Student's t-test using Microsoft Excel.
GOBO analysis
Gene expression-based Outcome for Breast cancer Online database was used to identify prognostic validation of RAD51C expression and ERα status of breast tumors collected from pooled breast cancer data sets http://co.bmc.lu.se/gobo/. The Gene Set Analysis (GSA) of tumors were further subdivided based on RAD51C expression with gray representing low expressing, red representing intermediate expressing and blue representing high expressing.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Funding was provided by NIH - CA151112; Atol Charitable Trust; LAM Foundation -098P0113; American Cancer Society - RSG-13-287-01-TBE.
ORCID
Rachel S. Salamon http://orcid.org/0000-0001-6548-2168
Subrata Manna http://orcid.org/0000-0001-9534-3935
References
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nature08467
- Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 2004; 4:665-76; PMID:15343273; http://dx.doi.org/10.1038/nrc1431
- Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 2001; 27:247-54; PMID:11242102; http://dx.doi.org/10.1038/85798
- Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002; 108:171-82; PMID:11832208; http://dx.doi.org/10.1016/S0092-8674(02)00615-3
- Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 2000; 408:429-32; PMID:11100717; http://dx.doi.org/10.1038/35044000
- Ogawa T, Yu X, Shinohara A, Egelman EH. Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science 1993; 259:1896-9; PMID:8456314; http://dx.doi.org/10.1126/science.8456314
- Thacker J. The RAD51 gene family, genetic instability and cancer. Cancer letters 2005; 219:125-35; PMID:15723711; http://dx.doi.org/10.1016/j.canlet.2004.08.018
- Lin Z, Kong H, Nei M, Ma H. Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc Natl Acad Sci U S A 2006; 103:10328-33; PMID:16798872; http://dx.doi.org/10.1073/pnas.0604232103
- Albala JS, Thelen MP, Prange C, Fan W, Christensen M, Thompson LH, Lennon GG. Identification of a novel human RAD51 homolog, RAD51B. Genomics 1997; 46:476-9; PMID:9441753; http://dx.doi.org/10.1006/geno.1997.5062
- Cartwright R, Tambini CE, Simpson PJ, Thacker J. The XRCC2 DNA repair gene from human and mouse encodes a novel member of the recA/RAD51 family. Nucleic Acids Res 1998; 26:3084-9; PMID:9628903; http://dx.doi.org/10.1093/nar/26.13.3084
- Dosanjh MK, Collins DW, Fan W, Lennon GG, Albala JS, Shen Z, Schild D. Isolation and characterization of RAD51C, a new human member of the RAD51 family of related genes. Nucleic Acids Res 1998; 26:1179-84; PMID:9469824; http://dx.doi.org/10.1093/nar/26.5.1179
- Pittman DL, Weinberg LR, Schimenti JC. Identification, characterization, and genetic mapping of Rad51d, a new mouse and human RAD51/RecA-related gene. Genomics 1998; 49:103-11; PMID:9570954; http://dx.doi.org/10.1006/geno.1998.5226
- Masson JY, Tarsounas MC, Stasiak AZ, Stasiak A, Shah R, McIlwraith MJ, Benson FE, West SC. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Gen Dev 2001; 15:3296-307; PMID:11751635; http://dx.doi.org/10.1101/gad.947001
- French CA, Tambini CE, Thacker J. Identification of functional domains in the RAD51L2 (RAD51C) protein and its requirement for gene conversion. J Biol Chem 2003; 278:45445-50; PMID:12966089; http://dx.doi.org/10.1074/jbc.M308621200
- Kuznetsov SG, Haines DC, Martin BK, Sharan SK. Loss of Rad51c leads to embryonic lethality and modulation of Trp53-dependent tumorigenesis in mice. Cancer Res 2009; 69:863-72; PMID:19155299; http://dx.doi.org/10.1158/0008-5472.CAN-08-3057
- Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci U S A 1996; 93:6236-40; PMID:8692798; http://dx.doi.org/10.1073/pnas.93.13.6236
- Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, Brookman KW, Siciliano MJ, Walter CA, Fan W, et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell 1998; 1:783-93; PMID:9660962; http://dx.doi.org/10.1016/S1097-2765(00)80078-7
- Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 2001; 21:2858-66; PMID:11283264; http://dx.doi.org/10.1128/MCB.21.8.2858-2866.2001
- French CA, Masson JY, Griffin CS, O'Regan P, West SC, Thacker J. Role of mammalian RAD51L2 (RAD51C) in recombination and genetic stability. J Biol Chem 2002; 277:19322-30; PMID:11912211; http://dx.doi.org/10.1074/jbc.M201402200
- Gruver AM, Miller KA, Rajesh C, Smiraldo PG, Kaliyaperumal S, Balder R, Stiles KM, Albala JS, Pittman DL. The ATPase motif in RAD51D is required for resistance to DNA interstrand crosslinking agents and interaction with RAD51C. Mutagenesis 2005; 20:433-40; PMID:16236763; http://dx.doi.org/10.1093/mutage/gei059
- Godthelp BC, Wiegant WW, van Duijn-Goedhart A, Scharer OD, van Buul PP, Kanaar R, Zdzienicka MZ. Mammalian Rad51C contributes to DNA cross-link resistance, sister chromatid cohesion and genomic stability. Nucleic Acids Res 2002; 30:2172-82; PMID:12000837; http://dx.doi.org/10.1093/nar/30.10.2172
- Yoshihara T, Ishida M, Kinomura A, Katsura M, Tsuruga T, Tashiro S, Asahara T, Miyagawa K. XRCC3 deficiency results in a defect in recombination and increased endoreduplication in human cells. EMBO J 2004; 23:670-80; PMID:14749735; http://dx.doi.org/10.1038/sj.emboj.7600087
- Renglin Lindh A, Schultz N, Saleh-Gohari N, Helleday T. RAD51C (RAD51L2) is involved in maintaining centrosome number in mitosis. Cytogenet Genome Res 2007; 116:38-45; PMID:17268176; http://dx.doi.org/10.1159/000097416
- Brenneman MA, Weiss AE, Nickoloff JA, Chen DJ. XRCC3 is required for efficient repair of chromosome breaks by homologous recombination. Mutat Res 2000; 459:89-97; PMID:10725659; http://dx.doi.org/10.1016/S0921-8777(00)00002-1
- Cui X, Brenneman M, Meyne J, Oshimura M, Goodwin EH, Chen DJ. The XRCC2 and XRCC3 repair genes are required for chromosome stability in mammalian cells. Mutat Res 1999; 434:75-88; PMID:10422536; http://dx.doi.org/10.1016/S0921-8777(99)00010-5
- Wiegmans AP, Al-Ejeh F, Chee N, Yap PY, Gorski JJ, Da Silva L, Bolderson E, Chenevix-Trench G, Anderson R, Simpson PT, et al. Rad51 supports triple negative breast cancer metastasis. Oncotarget 2014; 5:3261-72; PMID:24811120; http://dx.doi.org/10.18632/oncotarget.1923
- Wiegmans AP, Miranda M, Wen SW, Al-Ejeh F, Moller A. RAD51 inhibition in triple negative breast cancer cells is challenged by compensatory survival signaling and requires rational combination therapy. Oncotarget 2016 [epub ahead of print]; PMID:27507046
- Bric A, Miething C, Bialucha CU, Scuoppo C, Zender L, Krasnitz A, Xuan Z, Zuber J, Wigler M, Hicks J, et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell 2009; 16:324-35; PMID:19800577; http://dx.doi.org/10.1016/j.ccr.2009.08.015
- Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L, Schaal H, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 2010; 42:410-4; PMID:20400964; http://dx.doi.org/10.1038/ng.569
- Romero A, Perez-Segura P, Tosar A, Garcia-Saenz JA, Diaz-Rubio E, Caldes T, de la Hoya M. A HRM-based screening method detects RAD51C germ-line deleterious mutations in Spanish breast and ovarian cancer families. Breast Cancer Res Treat 2011; 129:939-46; PMID:21537932; http://dx.doi.org/10.1007/s10549-011-1543-x
- Vuorela M, Pylkas K, Hartikainen JM, Sundfeldt K, Lindblom A, von Wachenfeldt Wappling A, Haanpaa M, Puistola U, Rosengren A, Anttila M, et al. Further evidence for the contribution of the RAD51C gene in hereditary breast and ovarian cancer susceptibility. Breast Cancer Res Treat 2011; 130:1003-10; PMID:21750962; http://dx.doi.org/10.1007/s10549-011-1677-x
- Barlund M, Monni O, Kononen J, Cornelison R, Torhorst J, Sauter G, Kallioniemi O-P, Kallioniemi A. Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer Res 2000; 60:5340-4; PMID:11034067
- Sinclair CS, Rowley M, Naderi A, Couch FJ. The 17q23 amplicon and breast cancer. Breast Cancer Res Treat 2003; 78:313-22; PMID:12755490; http://dx.doi.org/10.1023/A:1023081624133
- Wu GJ, Sinclair CS, Paape J, Ingle JN, Roche PC, James CD, Couch FJ. 17q23 amplifications in breast cancer involve the PAT1, RAD51C, PS6K, and SIGma1B genes. Cancer Res 2000; 60:5371-5; PMID:11034073
- Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res 1999; 59:3547-51; PMID:10446958
- Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997; 386:804-10; PMID:9126738; http://dx.doi.org/10.1038/386804a0
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997; 88:265-75; PMID:9008167; http://dx.doi.org/10.1016/S0092-8674(00)81847-4
- Bennett BT, Knight KL. Cellular localization of human Rad51C and regulation of ubiquitin-mediated proteolysis of Rad51. J Cell Biochem 2005; 96:1095-109; PMID:16215984; http://dx.doi.org/10.1002/jcb.20640
- Gildemeister OS, Sage JM, Knight KL. Cellular redistribution of Rad51 in response to DNA damage: novel role for Rad51C. J Biol Chem 2009; 284:31945-52; PMID:19783859; http://dx.doi.org/10.1074/jbc.M109.024646
- Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol 2011; 22:898-905; PMID:21821141; http://dx.doi.org/10.1016/j.semcdb.2011.07.019
- Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet 2010; 42:406-9; PMID:20400963; http://dx.doi.org/10.1038/ng.570
- Badie S, Liao C, Thanasoula M, Barber P, Hill MA, Tarsounas M. RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J Cell Biol 2009; 185:587-600; PMID:19451272; http://dx.doi.org/10.1083/jcb.200811079
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003; 3:421-9; PMID:12781359; http://dx.doi.org/10.1016/S1535-6108(03)00110-7
- Nevanlinna H, Bartek J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 2006; 25:5912-9; PMID:16998506; http://dx.doi.org/10.1038/sj.onc.1209877
- Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 2002; 31:55-9; PMID:11967536; http://dx.doi.org/10.1038/ng879
- Cybulski C, Gorski B, Huzarski T, Masojc B, Mierzejewski M, Debniak T, Teodorczyk U, Byrski T, Gronwald J, Matyjasik J, et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet 2004; 75:1131-5; PMID:15492928; http://dx.doi.org/10.1086/426403
- Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin? Nat Rev Cancer 2007; 7:925-36; PMID:18004398; http://dx.doi.org/10.1038/nrc2251
- Zhuang J, Zhang J, Willers H, Wang H, Chung JH, van Gent DC, Hallahan DE, Powell SN, Xia F. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res 2006; 66:1401-8; PMID:16452195; http://dx.doi.org/10.1158/0008-5472.CAN-05-3278
- Williamson LM, Lees-Miller SP. Estrogen receptor alpha-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 2011; 32:279-85; PMID:21112959; http://dx.doi.org/10.1093/carcin/bgq255
- Hsu PY, Hsu HK, Lan X, Juan L, Yan PS, Labanowska J, Heerema N, Hsiao TH, Chiu YC, Chen Y, et al. Amplification of distant estrogen response elements deregulates target genes associated with tamoxifen resistance in breast cancer. Cancer Cell 2013; 24:197-212; PMID:23948299; http://dx.doi.org/10.1016/j.ccr.2013.07.007
- Maruani DM, Spiegel TN, Harris EN, Shachter AS, Unger HA, Herrero-Gonzalez S, Holz MK. Estrogenic regulation of S6K1 expression creates a positive regulatory loop in control of breast cancer cell proliferation. Oncogene 2012; 31:5073-80; PMID:22286763; http://dx.doi.org/10.1038/onc.2011.657
- Han HS, Yu E, Song JY, Park JY, Jang SJ, Choi J. The estrogen receptor alpha pathway induces oncogenic Wip1 phosphatase gene expression. Mol Cancer Res 2009; 7:713-23; PMID:19435816; http://dx.doi.org/10.1158/1541-7786.MCR-08-0247
- Bhat-Nakshatri P, Wang G, Collins NR, Thomson MJ, Geistlinger TR, Carroll JS, Brown M, Hammond S, Srour EF, Liu Y, et al. Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res 2009; 37:4850-61; PMID:19528081; http://dx.doi.org/10.1093/nar/gkp500
- Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, Nobel AB, van't Veer LJ, Perou CM. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med 2006; 355:560-9; PMID:16899776; http://dx.doi.org/10.1056/NEJMoa052933
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490:61-70; PMID:23000897; http://dx.doi.org/10.1038/nature11412
- Alayev A, Salamon RS, Berger SM, Schwartz NS, Cuesta R, Snyder RB, Holz MK. mTORC1 directly phosphorylates and activates ERalpha upon estrogen stimulation. Oncogene 2016; 35(27):3535–3543; PMID:26522726
- Tumiati M, Munne PM, Edgren H, Eldfors S, Hemmes A, Kuznetsov SG. Rad51c- and Trp53-double-mutant mouse model reveals common features of homologous recombination-deficient breast cancers. Oncogene 2016; 35:4601-10; PMID:26820992; http://dx.doi.org/10.1038/onc.2015.528
- Gevensleben H, Bossung V, Meindl A, Wappenschmidt B, de Gregorio N, Osorio A, Romero A, Buettner R, Markiefka B, Schmutzler RK. Pathological features of breast and ovarian cancers in RAD51C germline mutation carriers. Virchows Arch 2014; 465:365-9; PMID:24993905; http://dx.doi.org/10.1007/s00428-014-1619-1
- Woditschka S, Evans L, Duchnowska R, Reed LT, Palmieri D, Qian Y, Badve S, Sledge G, Jr., Gril B, Aladjem MI, et al. DNA double-strand break repair genes and oxidative damage in brain metastasis of breast cancer. J Natl Cancer Inst 2014; 106(7); PMID:24948741; http://dx.doi.org/10.1093/jnci/dju145
- Yata K, Bleuyard JY, Nakato R, Ralf C, Katou Y, Schwab RA, Niedzwiedz W, Shirahige K, Esashi F. BRCA2 coordinates the activities of cell-cycle kinases to promote genome stability. Cell Rep 2014; 7:1547-59; PMID:24835992; http://dx.doi.org/10.1016/j.celrep.2014.04.023
- Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006; 25:5864-74; PMID:16998501; http://dx.doi.org/10.1038/sj.onc.1209874
- Jordan VC. The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer. Endocr Relat Cancer 2015; 22:R1-31; PMID:25339261; http://dx.doi.org/10.1530/ERC-14-0448
- Min A, Im SA, Yoon YK, Song SH, Nam HJ, Hur HS, Kim HP, Lee KH, Han SW, Oh DY, et al. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol Cancer Ther 2013; 12:865-77; PMID:23512992; http://dx.doi.org/10.1158/1535-7163.MCT-12-0950
- Kim H, Tarhuni A, Abd Elmageed ZY, Boulares AH. Poly(ADP-ribose) polymerase as a novel regulator of 17beta-estradiol-induced cell growth through a control of the estrogen receptor/IGF-1 receptor/PDZK1 axis. J Translat Med 2015; 13:233; PMID:26183824; http://dx.doi.org/10.1186/s12967-015-0589-7
- Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ, Holz MK. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J Biol Chem 2009; 284:6361-9; PMID:19112174; http://dx.doi.org/10.1074/jbc.M807532200
- Alayev A, Sun Y, Snyder RB, Berger SM, Yu JJ, Holz MK. Resveratrol prevents rapamycin-induced upregulation of autophagy and selectively induces apoptosis in TSC2-deficient cells. Cell cycle 2014; 13:371-82; PMID:24304514; http://dx.doi.org/10.4161/cc.27355