
ABSTRACT
Spindle poisons elicit various cellular responses following metaphase arrest, but how they relate to long-term clonogenicity has remained unclear. We prepared several HeLa lines in which the canonical apoptosis pathway was attenuated, and compared their acute responses to paclitaxel, as well as long-term fate, with the parental line. Three-nanomolar paclitaxel induced brief metaphase arrest (<5 h) often followed by aberrant mitosis, and about 90% of the cells of each line had lost their clonogenicity after 48 h of the treatment. A combination of the same concentration of paclitaxel with the kinesin-5 inhibitor, S-trityl-L-cysteine (STLC), at 1 µM led to much longer arrest (∼20 h) and predominance of subsequent line-specific responses: mitochondrial outer membrane permeabilization (MOMP) in the apoptosis-prone line, or mitotic slippage without obvious MOMP in the apoptosis-reluctant lines. In spite of this, combination with STLC did not lead to a marked difference in clonogenicity between the apoptosis-prone and -reluctant lines, and intriguingly resulted in slightly better clonogenicity than that of cells treated with 3 nM paclitaxel alone. This indicates that changes in the short-term response within 3 possible scenarios — acute MOMP, mitotic slippage or aberrant mitosis ― has only a weak impact on clonogenicity. Our results suggest that once cells have committed to slippage or aberrant mitosis they eventually undergo proliferative death irrespective of canonical apoptosis or p53 function. Consistent with this, cells with irregular DNA contents originating from mitotic slippage or aberrant mitosis were mostly eliminated from the population within several rounds of division after the drug treatment.
Introduction
Anomaly of the mitotic spindle induced by taxanes or vinca alkaloids leads to activation of the spindle assembly checkpoint (SAC) with concomitant metaphase arrest, and frequently to subsequent loss of cell clonogenicity. Although such proliferative cell death may involve both apoptotic and non-apoptotic processes, the relationship between them, their relative importance, and the underlying molecular mechanisms are still unclear.
Previous studies have shown that cells under tight metaphase arrest eventually either die through apoptosis or undergo mitotic slippage, i.e, exit from mitosis into interphase without cell division. A competing networks model has predicted that immediate fate following metaphase arrest is determined by which threshold is satisfied earlier, accumulation of cell death signals for apoptosis, or a decrease of the cyclin B1 level for slippage.Citation1,2 Although decay of MCL1 during mitotic arrest, possibly due to absence of transcription, is implicated as an apoptosis trigger in this context, the contribution of other pro-survival factor(s) is also suspected. Death signals thus generated cause activation of the caspase-dependent authentic apoptosis pathway through mitochondrial outer membrane permeabilization (MOMP).Citation3-6 On the other hand, in some cases, slipped cells undergo delayed apoptosis involving multinucleation,Citation7 whereas in others they are subjected to G1 arrest initiated by either DNA damage or non-damage stresses,Citation8-10 both of which are reported to be dependent on p53 and its downstream targets such as p21. The long-term fate of the slipped cells is obscure, especially when p53 function is compromised.
Under the situation where mitotic arrest is not sufficiently robust in spite of the presence of a spindle defect, cells may exhibit another scenario whereby the SAC gets turned off stochastically after temporary metaphase arrest, and the cells commit to aberrant mitosis, often giving rise to aneuploid daughter cells.Citation11-13 When such daughter cells are devoid of genetic information essential for survival and proliferation, they may die in the absence of active elimination mechanisms, even if they avoid an acute form of cell death known as mitotic catastrophe. If cells happen to inherit sufficient but imbalanced genetic material, they may continue to divide and could become a source of tumor initiation or malignancy. This classical hypothesis, originally proposed by Theodor Boveri, has never been fully verified.Citation14
Some tumors show innate or acquired resistance to taxane-based chemotherapeutics, and this has been partly attributed to a low propensity for apoptosis.Citation15,16 If apoptosis is the single leading cause of proliferative cell death after taxane treatment, its absence would lead to not only drug resistance but also a significant fraction of aneuploid or polyploid cells among clonogenic survivors. In contrast, if aneuploid/polyploid cells are subjected to an elimination mechanism other than apoptosis, the effect of apoptosis-reluctance would be much less obvious. The latter possibility is apparently consistent with a long-standing claim that loss of apoptosis barely affects clonogenicity after cells of non-hematological origin have been irradiated or treated with various cytotoxic chemicals.Citation17,18
To directly assess how impairment of the canonical apoptosis pathway affects the short-term response after metaphase arrest as well as the long-term clonogenicity of cancer cells treated with a spindle poison, we prepared a series of apoptosis-reluctant HeLa cell lines by either stable overexpression of BCL2 and BCLxL or simultaneous knockout of the BAX and BAK1 genes, and investigated their responses to different doses of paclitaxel. In HeLa cells, p53 is considered to be inactivated by papillomavirus protein E6,Citation19,20 and therefore pathways that are dependent on p53 function may not be functional. To correlate immediate cellular responses with clonogenicity, we utilized relatively low concentrations of paclitaxel at which clonogenic survival is within an relevant range (5–20%), as opposed to most studies focusing on acute cellular responses alone employing much higher concentrations. The results we obtained suggest that while inactivation of canonical apoptosis dramatically changes the spectrum of cellular short-term fates, it barely affects the rate of proliferative cell death. Intriguingly, a combination of 2 different spindle poisons induced apoptosis at a higher rate, as well as being slightly less lethal to apoptosis-prone cells under certain conditions. The possible reasons for this are discussed.
Materials and methods
Cell culture and media
All cells were maintained in a humidified atmosphere with 5% CO2 at 37°C. HeLa and HMVII cells were obtained from the Cell Resource Center for Biomedical Research at Tohoku University and cultured in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% FBS. 293T cells were obtained from RIKEN BRC Cell Bank and cultured in DMEM medium (Thermo Fisher Scientific) supplemented with 10% FBS.
Plasmids
A set of plasmids for lentivirus-mediated gene expression pCAG-HIVgp, pCMV-VSV-G-RSV-Rev and pCSII-EF-MCS were obtained from Dr. H. Miyoshi at RIKEN Tsukuba Institute. A plasmid for expression of G2/M reporter hGem(GMNN)-Venus (a component of Fucci2) was obtained from Dr. A. Miyawaki at RIKEN Brain Science Institute. For MOMP visualization, a DNA fragment encoding the mitochondrial translocation signal of human DIABLO (AA1-55) was RT-PCR amplified with SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) and KOD Plus DNA polymerase (Toyobo) from total RNA of the human melanoma cell line SK-MEL-28 (our laboratory stock). The obtained fragment was cloned into pCSII-EF-MCS via fusion with the mCherry gene (pSY329). For ectopic expression of BCLxL and BCL2, PCR-amplified cDNA fragments fused with 3xHA or 3xFLAG tag sequence were cloned into pCSII-EF-MCS (pSY327 and pSY324). For Crispr/CAS9-mediated genome editing, gRNAs were designed for knock-out of the 4th exon of human BAX or the 3rd and 4th exons of human BAK1 (Fig. S2), and the corresponding short double-stranded DNA fragments were inserted at the BbsI sites of pX330 purchased from Addgene (Fig. S2, pSY332 and pSY333 for BAX, and pSY335 and pSY336 for BAK1).
Crispr/CAS9-mediated gene knockout
To simplify screening for gene deletion, a pair of guide RNA expression plasmids targeting 2 adjacent sites (about a few hundred bases apart) were used for a single gene-knock out attempt. Five million HeLa cells were co-transfected with a mixture of 5 µg each of 2 gRNA/CAS9 expression plasmids and 1 µg of a puromycin-resistance marker-containing plasmid using a GenePulser Xcell electroporator (Bio-Rad). To increase the knock-out efficiency, cells were cultured in medium containing 0.5 µg/ml puromycin for 2 days and about 2 dozen clones were obtained by limiting dilution and cultured in fresh medium. Genomic DNA from each clone was screened by PCR for deletion, and gene inactivation was confirmed by immunoblotting.
Lentiviral vector-mediated ectopic gene expression
A replication-defective, self-inactivating lentivirus for ectopic gene expression was prepared essentially as described (http://cfm.brc.riken.jp/lentiviral-vectors/protocols/). Briefly, 17 µg of pCSII-EF-MCS-based plasmids containing DIABLO-mCherry, GMNN-Venus, BCLxL-3xHA or BCL2-3xFLAG was co-transfected with 10 µg each of the packaging plasmid pCAG-HIVgp and the VSV-G/Rev-expressing plasmid pCMV-VSV-G-RSV-Rev into 293T cells using the calcium phosphate co-precipitation method. The medium was replaced after 16 h of transfection and the cells were cultured for a further 48 h. Viral particles were concentrated from the recovered medium using Lenti-X-Concentrator (#631231, Takara Bio) and the titer was estimated with Lenti-X-GoStix (#631243, Takara Bio). HeLa GI23 cells were transduced at a MOI of 10 and stable clones were obtained by limiting dilution.
Cell viability assay
Cell survival immediately after drug treatment was evaluated with the water-soluble tetrazolium salts (WST) assay using a Cell Counting Kit-8 (Dojindo Laboratories). Cells were seeded into 96-well plates (2 × 103 cells/well) and cultured for 16–24 h prior to treatment with paclitaxel and STLC. After the treatment, the medium in each well was replaced with 100 µl of drug-free fresh medium and 10 μl of Cell Counting Kit-8 solution, incubated for an additional 1–2 h, and the absorbance at 450 nm was measured using a Multiskan Spectrum spectrophotometer (Thermo Fisher Scientific). To evaluate bulk growth, cells were seeded into 24-well plates (2 × 103-2 × 104 cells /well), treated with the drug for 48 h and cultured for a further 5 days in fresh medium. The cells were then fixed by adding a 1/10 volume of 37% formaldehyde solution, stained with 0.5% crystal violet 20% methanol, and photographed. To measure clonogenicity, 5 × 105 cells were seeded into a 90-mm dish and treated with the drug for 48 h. Cells including those that had detached from the dish during the treatment were harvested, counted, and 200 to 2,000 cells were replated into a 60-mm dish in triplicate. After 7 to 10 days of culture, the cells were fixed and stained, and colonies were counted.
Protein preparation and immunoblotting
After appropriate treatment, 1–5×106 cells were washed twice with ice-chilled PBS, fixed with 10% TCA in saline for 1 h on ice, and then scraped off into a tube. The cell pellet was washed once with deionized water and lysed in 9 M urea, 2% Triton X-100 and 1% DTT. Protein concentration was measured with a BCA protein assay kit (Merck Millipore Corporation) before addition of DTT. Approximately 20–30 µg of protein per lane was electrophoresed on 10% SDS-PAGE gel for 30 min at 200 V and then transferred onto polyvinylidene fluoride transfer membranes (Pall Corporation). The membranes were blocked with 5% low-fat dried milk (Morinaga Milk Industry) in 1×TBS-T for 1 h at room temperature and then immuno-reacted with an appropriate primary antibody overnight at 4°C and subsequently with HRP-conjugated secondary antibodies (GE Healthcare Life Sciences) with 1:4000 dilution for 1 h at room temperature. Signals were visualized with ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences) and ChemiDoc XRS (Bio-Rad Laboratories). Intensity of the signals was quantified using ImageJ/Fiji softwareCitation21 and normalized against the α-tubulin or GAPDH signal. The following primary antibodies were used at the indicated dilutions: mouse anti-FLAG tag (F3165, Sigma Aldrich, 1:1000), rat anti-hemagglutinin tag (11867423001, Roche Diagnostics, 1:1000), mouse anti-α-tubulin (T5168, Sigma Aldrich, 1:1000), mouseanti-BCL2 (05-729, Merck Millipore Corporation, 1:250), rabbit anti-BCLxL (#2764S, Cell Signaling Technology, 1:1000), rabbit anti-BAX (#2772S, Cell Signaling Technology, 1:1000), rabbit anti-BAK1 (#3814S, Cell Signaling Technology, 1:1000), rabbit anti-PARP (#9542S, Cell Signaling Technology, 1:1000) and rabbit anti-GAPDH (#5174S, Cell Signaling Technology, 1:1000). Electrophoresis and immunoblotting were repeated at least 3 times with independent sample preparations and confirmed to be reproducible.
Time-lapse microscopy
Ten thousands cells were seeded in each of 4 compartments of CELLview™ glass-bottom dishes (Greiner Bio-One GmbH) and cultured for 16–24 h. Observation was started immediately after drug addition using a Keyence BZ-9000 fluorescence microscope (Keyence) fitted with appropriate filter sets and a stage incubator INUG2-KI2 (Tokai Hit). To minimize phototoxicity, the time-lapse interval was set to 1 h and images were captured until 71 h after drug addition. For each time-point, fifteen 2.5 µm-interval Z-sections were taken and the section with the best focus for each fluorescence channel was picked up semi-automatically using ImageJ/Fiji macro language.
Flow cytometric analysis of DNA content
A million ethanol-fixed cells were rehydrated with PBS, resuspended in 1 ml of PBS containing 0.1% TritonX-100, 100 µg/ml RNase A and 10 µg/ml propidium iodide, and incubated at 37°C for 30 min. The cells were briefly sonicated with a Branson Sonifier 150 (Emerson Industrial Automation) to remove clumps and analyzed with a BD FACSCalibur™ (BD Bioscience).
Immunofluorescence microscopy
Twenty thousand cells were seeded onto a pre-washed and poly-L-lysine-coated 18 mm-square coverslip and allowed to attach at 37°C overnight. The cells were then fixed with acetone/methanol at −20°C for 20 min. Most of the Venus/mCherry fluorescence was quenched under this fixation condition. The cells were blocked with PBS containing 2% low fat milk, 0.2% BSA, and 0.2% Tween-20 at room temperature for 30 min, treated with rat anti-HA antibody (11867423001, Roche Diagnostics) at 1:1000 dilution in PBS containing 2% BSA for 2 h, and subsequently with Alexa 488-conjugated anti-Rat IgG goat antibody (Thermo Fisher Scientific) at 1:1000 dilution for 2 h. After washing, the coverslips were mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific).
Results
Construction of apoptosis-prone or -reluctant HeLa cell lines with MOMP and cell cycle reporters and validation of their phenotype
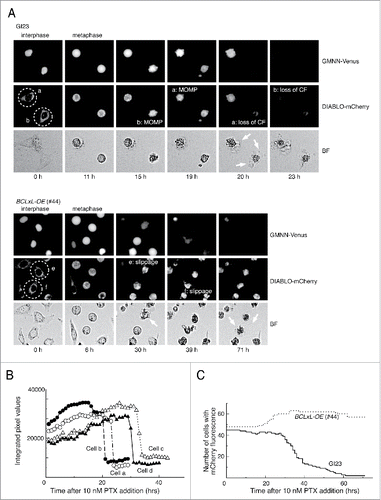
To monitor cell cycle progression and paclitaxel-induced apoptosis in living cells, we first established a HeLa-based cell line that stably co-expresses the GMNN(Geminin)-VenusCitation22,23 and DIABLO(SMAC)-mCherryCitation24 fusion proteins, which function as reporters of the G2/M cell cycle phase and MOMP, respectively (HeLa-GI23 line, Fig. S1). Since the fusion proteins lack domains essential for the native functions of GMNN and DIABLO, their expression would not disturb DNA replication or apoptosis. We further constructed several apoptosis-reluctant lines from GI23: line #44 over-expressing BCLxL-HA, line #83 over-expressing both BCLxL-HA and BCL2-FLAG, and lines #69, #88 and #89 in which both the BAX and BAK1 genes are deleted (Fig. S2, S3). Treatment of asynchronously growing GI23 cells with 10 nM paclitaxel led to metaphase arrest and a subsequent change in the subcellular distribution of mCherry fluorescence, as a sign of MOMP (). The MOMP was followed by an abrupt decrease in the intensity of cellular mCherry fluorescence within several hours, probably due to loss of plasma membrane integrity ( and ). In the time-lapse microscopy experiments conducted subsequently, in which direct judgment of MOMP was often difficult, loss of cellular mCherry fluorescence was used as index of apoptosis instead. Most of the GI23 cells (>90%) underwent MOMP or fluorescence loss within 60 h after addition of 10 nM paclitaxel (). In contrast, for most of the line #44 cells, mCherry fluorescence was maintained until the end of observation with no clear sign of MOMP ( and ). Lines #83, #69, #88 and #89 also showed resistance to loss of mCherry fluorescence upon treatment with 10 nM paclitaxel (see below). Under the same conditions, efficient cleavage of PARP took place in the GI23 line, whereas its kinetics were notably compromised in the apoptosis-reluctant lines (Fig. S4). Nevertheless, some residual cleavage was observed in these lines, possibly representing an atypical cell death process that does not depend on BAX/BAK1. Consistent with this, apoptosis-reluctant line #44 showed extensive blebbing and a shrunken appearance after slippage ( and ), as is often seen in apoptotic cells.
Figure 1. Typical responses of the prepared HeLa cell lines to paclitaxel. (A) Time-lapse observation of MOMP-prone (GI23) and MOMP-reluctant (#44) HeLa cells treated with 10 nM paclitaxel. Contrast of bright field images (BF) is computer-enhanced. The time after drug addition is indicated below the panels. Exit from mitosis was judged by the loss of GMMN-Venus fluorescence. Cells a and b (encircled with dotted lines) of GI23 terminated metaphase arrest with MOMP, followed by loss of mCherry fluorescence (CF) and blebbing (white arrows). Cells e and f of line #44 slipped out of metaphase arrest and underwent blebbing (white arrows) while retaining mCherry fluorescence until the end of observation. (B) Switch-like loss of mCherry fluorescence in each of 4 GI23 cells treated with 10 nM paclitaxel. Cells a and b are identical to those shown in (A). (C). The number of cells retaining mCherry fluorescence is plotted against time after addition of 10 nM paclitaxel. All cells in a single microscopic field were counted for each respective cell line. The slight increase in the number of #44 cells is due to mitosis of the minor fraction.
Time-lapse microscopy of -cells treated with 2 different paclitaxel concentrations
To evaluate the short-term responses of the cells in a more quantitative manner, we classified each of the first mitoses of the 4 cell lines in the presence of paclitaxel into several categories according to how they ended, and plotted the duration of metaphase arrest (). As described above, essentially all GI23 cells were arrested in the first metaphase for 19 ± 6.0 h (n = 101) after addition of 10 nM paclitaxel, and more than 90% (93/101) exited mitosis (as judged by disappearance of GMNN-Venus fluorescence) with MOMP. In contrast, although the BCLxL- and BCL2-overexpressing cell (line #83) similarly showed arrest in the first metaphase for 22 ± 6.8 h (n = 57), most of the cells (54/57) exited from the arrest by mitotic slippage without showing MOMP or cell division. We noticed that during mitotic arrest of cells destined for slippage, cellular GMNN-Venus fluorescence increased to a certain level then declined slowly (Fig S5), being reminiscent of the kinetics of GFP-tagged cyclin B in mitotic slippage.Citation1
Figure 2. Effect of paclitaxel treatment on mitosis and survival of apoptosis-prone and -reluctant cell lines. (A) Duration of mitotic arrest of the 4 cell lines and their behavior on mitotic exit after treatment with 3 and 10 nM paclitaxel. Horizontal lines represent mitotic arrest of individual cells sorted by the type of behavior on mitotic exit and the time of arrest onset. Black, (apparently) symmetric division; red, MOMP during arrest; orange, exit with no division (mitotic slippage); green, asymmetric division; blue, ternary division; purple, miscellaneous. (B) Cell survival immediately after paclitaxel treatment for 48 h measured using the WST assay (see Materials and Methods). (C) Colony-forming ability after paclitaxel treatment for 48 h. The results are shown as mean ± SEM (n = 3).
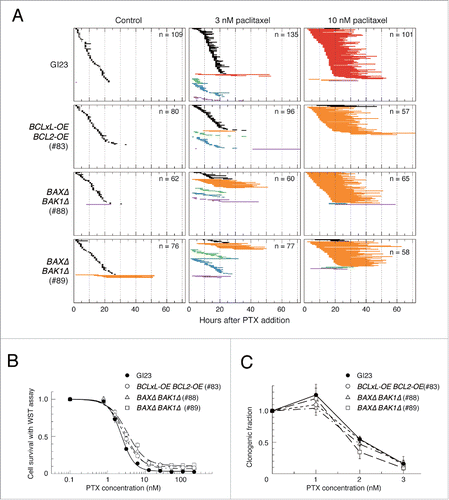
When treated with 3 nM paclitaxel, the GI23 cells were arrested in metaphase for a much shorter period (3.5 ± 4.4 h, n = 135). Most of the cells exited from mitosis with either apparently symmetrical (87/135), asymmetrical (9/135) or multipolar divisions (28/135, see Fig. S6 for examples of asymmetric and multipolar divisions) and only a minor population underwent MOMP (9/135). A high incidence of multipolar division in the presence of low paclitaxel concentrations is consistent with previous studies.Citation11,25,26 Most of the line #83 cells also terminated a short duration of metaphase arrest with either apparently normal or aberrant (including multipolar) cell division, while MOMP was almost never observed. The BAXΔ BAK1Δ cells (lines #88 and 89) responded to 3 nM paclitaxel similarly with a slightly higher incidence of mitotic slippage ().
Survival of cells treated with paclitaxel
To see whether apoptosis-reluctance leads to better survival after paclitaxel treatment, we treated the cells with 1 to 200 nM paclitaxel for 48 h and measured the number of living cells immediately after the treatment using the water-soluble tetrazolium salt (WST) assay. In spite of a substantial difference in the MOMP induction rate at high paclitaxel concentrations, BCLxL/BCL2-overexpressing cells (line #83) or BAXΔ BAK1Δ cells (lines #88 and 89) were barely more resistant to paclitaxel than the parental GI23 cells (). Since the WST assay may not necessarily represent the number of clonogenic cells, we also measured the bulk growth and colony-forming ability of the cells. After paclitaxel treatment for 48 h followed by culture in fresh medium for several days, there was little difference in growth or colony-forming ability between GI23 cells and the apoptosis-reluctant lines #83, #88 and #89 ( and , 2 upper rows). In the colony formation assay, trypan blue-positive cells at the time of cell harvest comprised less than 10% of the total even at the highest paclitaxel concentration (data not shown), suggesting that acute cell death was not predominant. Although the line #69 BAXΔ BAK1Δ cells showed remarkably better bulk growth after paclitaxel (, 2 upper rows), this may not have resulted from their low propensity for apoptosis, since other apoptosis-reluctant lines did not show such resistant growth. We also noticed that the colony formation assay showed a reproducibly higher survival rate than the bulk growth assay, for which the drug treatment protocols are slightly different. This did not affect our conclusion.
Figure 3. Duration of mitotic arrest of the 4 cell lines and their behavior on mitotic exit after treatment with 1 µM STLC with or without 3 nM paclitaxel. Horizontal lines represent duration of arrest and type of behavior on mitotic exit, as in .
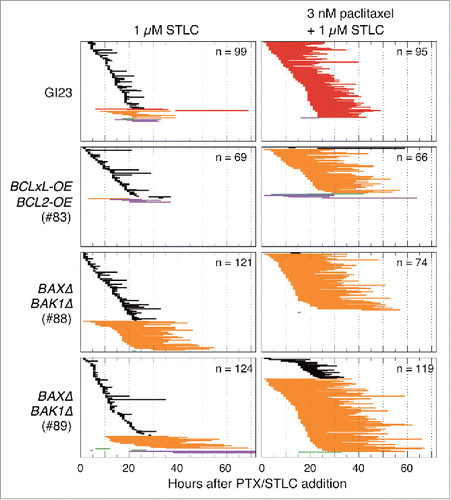
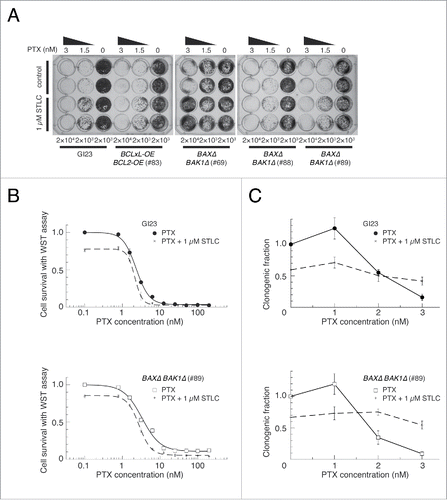
Figure 4. Effect of STLC on cell survival after paclitaxel treatment. (A) Bulk cell growth after drug treatment. Two thousand or 2 × 104 cells were plated into each well of 24-well plates, treated with appropriate drugs for 48 h, and incubated for a further 5–7 days in fresh medium. Upper two rows represent cells treated with paclitaxel alone and lower 2 rows represent cells treated with paclitaxel and 1 µM STLC. (B) Cell survival immediately after paclitaxel and STLC treatment for 48 h measured using the WST assay (see Materials and Methods). (C) Colony-forming ability after treatment with paclitaxel and STLC for 48 h. The results are shown as mean ± SEM (n = 3).
To confirm the results of the clonogenic assay, we mixed the GI23 cells and BCLxL-overexpressing cells (#44) at a ratio of 4:1 and treated the mixture with 1.5 nM paclitaxel for 48 h, followed by replating and culture for several days. Under the condition employed, cell growth was reduced to 10–20% after paclitaxel treatment. After immunofluorescence staining of BCLxL-HA, we saw no remarkable enrichment of BCLxL-overexpressing cells (from 110/597 to 61/618 in the paclitaxel-treated population versus from 110/597 to 106/707 in the control population), again suggesting that a low propensity for MOMP during mitotic arrest does not lead to better growth after paclitaxel treatment (Fig. S7).
Effect of kinesin-5 inhibitor on division of paclitaxel-treated cells
As described above, although about 90% of the cells (in the colony formation assay) or higher (in bulk growth assay) lost their clonogenicity after treatment with 3 nM paclitaxel for 48 h, only a minor fraction committed to MOMP during the first mitosis, even in the apoptosis-prone GI23 cells. A high frequency of aberrant cell division on exit from mitosis, or mitotic catastrophe, argues for its connection with cell death irrespective of MOMP, as has been suggested previously.Citation18 We next investigated the outcome of disturbance of mitotic catastrophe in the presence of a low concentration of paclitaxel. Reasoning that modest inhibition of centrosome separation would suppress aberrant nuclear division,Citation27 we investigated the response of the cells to a combination of paclitaxel at 3 nM and the kinesin-5 inhibitor STLC at 1 µMCitation28 using time-lapse microscopy (). Both the GI23 line and the apoptosis-reluctant lines were arrested in the first metaphase for a markedly longer duration (17 ± 5.1 and 22 ± 7.4 h for GI23 and line #83, respectively) than with either 3 nM paclitaxel (see above) or 1 µM STLC (4.4 ± 6.2 and 3.6 ± 4.7 h) alone. The prolonged metaphase arrest resulted in a notable decrease of aberrant mitosis and a compensatory increase of MOMP (in GI23) or mitotic slippage (in MOMP-reluctant lines), as was the case for the cells treated with 10 nM paclitaxel. Although treatment with 1 µM STLC alone led to a modest decrease in cell growth or clonogenicity, it slightly improved the clonogenicity of both MOMP-prone and MOMP-reluctant cells treated with paclitaxel (). While there was some variation in the degree of the suppressive effect among the cell lines (being rather weak in line #88), this tendency appeared to be general (). Note that addition of STLC induced more MOMP and less proliferative death in the paclitaxel-treated GI23 cells, again suggesting that MOMP-mediated acute apoptosis is not the single main cause of proliferative death in paclitaxel-treated cells.
Mutual suppression of clonogenicity loss between paclitaxel and STLC in Hela and apoptosis-reluctant melanoma cell lines
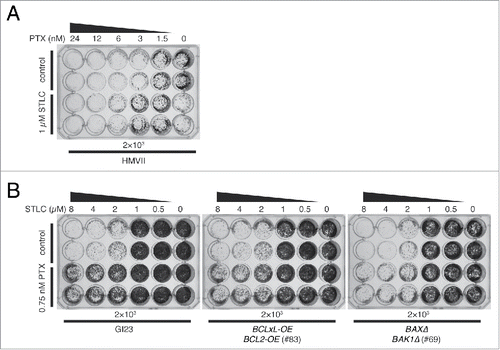
To examine the generality of STLC-mediated suppression of paclitaxel cytotoxicity, we treated the innately apoptosis-reluctant melenoma cell line HMVII with a combination of drugs. When the cells were exposed to increasing concentrations of paclitaxel alone for 48 h, proliferation was inhibited to less than 5% at a level of several nanomolar (, upper 2 rows). This was noteworthy, since HMVII is quite apoptosis-reluctant, and our previous study had shown that paclitaxel at 40 nM hardly caused PARP cleavage.Citation16 This again suggests that proliferative death in response to paclitaxel occurs mostly through a non-apoptotic process. We found that 1 µM STLC clearly suppressed the loss of clonogenicity due to paclitaxel at 3 to 6 nM (). Interestingly, whereas 1 µM STLC alone caused a slight decrease in proliferation of HMVII, whereas addition of 0.5 nM paclitaxel abrogated the effect of STLC suggesting that suppression occurred in the other direction also. We confirmed this phenomenon in a HeLa apoptosis-prone line and an apoptosis-reluctant line. In both GI23 and line #83, addition of 0.75 µM paclitaxel improved the proliferation of STLC-treated cells ().
Figure 5. Mutual suppressive effect of STLC and paclitaxel on cell killing. (A) Innately apoptosis-reluctant HMVII cells were treated with paclitaxel with (lower 2 rows) or without (upper 2 rows) 1 µM STLC, incubated for 7 days in fresh medium, and then stained. Note that the combination of 1 µM STLC and 0.5 nM paclitaxel resulted in better growth than with 1 µM STLC alone. (B) Growth of HeLa-based cell lines treated with an increasing dose of STLC with (lower 2 rows) or without (upper 2 rows) 0.75 nM paclitaxel. Addition of paclitaxel clearly suppressed cell killing by STLC.
DNA content of surviving cells after paclitaxel treatment
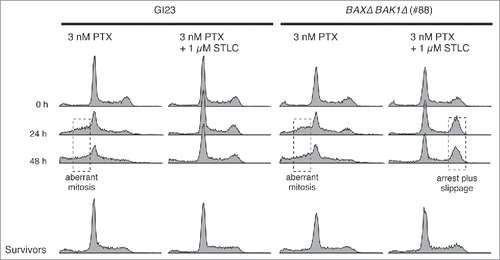
If a certain fraction of the cells that exited from mitosis by slippage or aberrant nuclear division retain their clonogenicity, we would expect to see polyploid or aneuploid cells in the surviving population after several rounds of cell division. Using flow cytometry, we measured the DNA content of the cells during paclitaxel treatment and in those that retained their clonogenicity after the treatment (). The untreated GI23 cells or the BAXΔ BAK1Δ cells (line #88) exhibited clear DNA peaks at G1 and G2/M. During treatment with 3 nM paclitaxel, the DNA distribution became blurred and a certain fraction of cells showed a DNA content lower than the G1 peak in both cell lines. Our results reproduced those of previous studies,Citation29 and were in good agreement with the occurrence of asymmetric or multipolar nuclear division observed by time-lapse microscopy under the same conditions as those described above. Note that this population is distinct from the so-called sub-G1 population usually regarded as an index of apoptosis. When the cells were treated with 3 nM paclitaxel and 1 µM STLC, cells with an intermediate DNA content were hardly evident, consistent with suppression of aberrant nuclear division. The fraction of cells with a G2/M DNA content was notably increased only in the apoptosis-reluctant line, probably due to accumulation of slipped cells. When the cells were cultured for 5–7 days in fresh medium after paclitaxel treatment for 48 h with or without STLC, essentially all irregularity of the DNA content disappeared and the proliferative cell population again showed a clear 2-peak distribution. Although a small karyotypic change would be below the resolution of flow cytometry, our results suggest that most cells with aberrant DNA contents would be eliminated during several rounds of the cell cycle after paclitaxel treatment, irrespective of canonical apoptosis.
Figure 6. DNA contents of apoptosis-prone and -reluctant cells during and after drug treatment. Surviving cells were cultured in fresh medium for 5–7 days after the treatment, then fixed and analyzed. Anomaly of the DNA content, probably due to aberrant mitosis or mitotic slippage, is indicated by dotted boxes.
Discussion
Consistent with the previous notion,Citation30,31 most of the paclitaxel-treated HeLa cells showed one out of 3 distinct outcomes in the present study: MOMP-mediated acute apoptosis or mitotic slippage after tight metaphase arrest, which typically occurred in the presence of a high paclitaxel concentration, or aberrant nuclear division after loose metaphase arrest in the presence of a low paclitaxel concentration. We showed that although inactivation of apoptosis or addition of STLC markedly altered the balance of the 3 immediate outcomes, it barely affected the clonogenicity of paclitaxel-treated cells. Together these results suggest that efficient elimination mechanisms other than acute apoptosis may operate in cells that have undergone aberrant mitosis or mitotic slippage, and that these may not depend on p53 function.
Currently, the details of such mechanisms are unclear. According to our preliminary observation, the slipped cells often did not accumulate GMNN-Venus fluorescence (data not shown), suggesting that permanent arrest or atypical cell death took place in the next G1 phase prior to G1/S transition, in accord with their apoptosis-like appearance. How this is able to occur in the absence of p53 will need to be explored in a future study. For daughter cells that originated from aberrant mitosis, most showed an increase of GMNN-Venus fluorescence, at least in the immediate cell cycle, suggesting that replication does proceed to a certain degree. Previous studies have shown that defective mitosis generates DNA damage, which then activates the p53-dependent G1/S checkpoint. We imagine that such DNA damage would activate the G2/M checkpoint in cells lacking functional p53. This possibility in the context of defective mitosis will also need be investigated.
Apart from the cell death or elimination mechanism, it is also unclear how a fraction of cells survived the spindle poison treatment, and more specifically why cells survived slightly better in the presence of 2 different spindle poisons. If aberrant mitosis or slippage necessarily leads to proliferative cell death as we have suggested, then surviving cells would be those that had avoided these outcomes. Although a competing networks model predicts that most cells either die or slip under tight metaphase arrest, such as that induced by a combination of drugs, a minor fraction of cells might escape both outcomes, at least during the limited time of treatment and restart the cell cycle, perhaps explaining the slightly better clonogenic survival. Zhu et al.Citation7 reported a similar suppressive effect of kinesin-5 inhibitor or knockdown of kinesin-5 on the cytotoxicity of paclitaxel, but they ascribed this to attenuation of delayed apoptosis, which seemingly contradicts our observation that STLC dramatically increased the MOMP rate in the apoptosis-prone line. This might be due to the difference between acute and delayed apoptosis, as well as due to differences in the concentrations of paclitaxel we adopted, being almost 2 orders of magnitude apart. It is possible that 2 apparently similar phenomena might occur through unrelated underlying mechanisms.
Although we used relatively mild paclitaxel treatment in the present study, survivors after the treatment represented only a minor fraction of the cell population, and it would be a challenge to detect their pivotal behavior using time-lapse microscopy. Nevertheless, such analysis in combination with appropriate molecular probes would provide valuable insight into their potential contribution to cancer malignancy or drug resistance.
Abbreviations
| MOMP | = | mitochondrial outer membrane permeabilization |
| SAC | = | spindle assembly checkpoint |
| STLC | = | S-trityl-L-cysteine |
| WST | = | water-soluble tetrazolium salts |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
supplementary.docx
Download MS Word (8.2 MB)Funding
This work was supported by JSPS KAKENHI Grant Number JP25460462.
Related Research Data
References
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008; 14:111-22; PMID:18656424; http://dx.doi.org/10.1016/j.ccr.2008.07.002
- Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 2007; 6:70-4; PMID:17245109; http://dx.doi.org/10.4161/cc.6.1.3682
- Wan L, Tan M, Yang J, Inuzuka H, Dai X, Wu T, Liu J, Shaik S, Chen G, Deng J, et al. APC(Cdc20) suppresses apoptosis through targeting bim for ubiquitination and destruction. Dev Cell 2014; 29:377-91; PMID:24871945; http://dx.doi.org/10.1016/j.devcel.2014.04.022
- Díaz-Martínez LA, Karamysheva ZN, Warrington R, Li B, Wei S, Xie X-J, Roth MG, Yu H. Genome-wide siRNA screen reveals coupling between mitotic apoptosis and adaptation. EMBO J 2014; 33:1960-76; PMID:25024437; http://dx.doi.org/10.15252/embj.201487826
- Wang P, Lindsay J, Owens TW, Mularczyk EJ, Warwood S, Foster F, Streuli CH, Brennan K, Gilmore AP. Phosphorylation of the proapoptotic BH3-only protein bid primes mitochondria for apoptosis during mitotic arrest. Cell Reports 2014; 7:661-71; PMID:24767991; http://dx.doi.org/10.1016/j.celrep.2014.03.050
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011; 471:110-4; PMID:21368834; http://dx.doi.org/10.1038/nature09779
- Zhu Y, Zhou Y, Shi J. Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle 2014; 13:1756-64; PMID:24694730; http://dx.doi.org/10.4161/cc.28672
- Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol 2010; 20:1666-71; PMID:20832310; http://dx.doi.org/10.1016/j.cub.2010.08.018
- Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell 2012; 23:567-76; PMID:22171325; http://dx.doi.org/10.1091/mbc.E11-09-0781
- Blagosklonny MV. Prolonged mitosis versus tetraploid checkpoint: how p53 measures the duration of mitosis. Cell Cycle 2006; 5:971-5; PMID:16687915; http://dx.doi.org/10.4161/cc.5.9.2711
- Chen J-G, Horwitz SB. Differential mitotic responses to microtubule-stabilizing and -destabilizing drugs. Cancer Res 2002; 62:1935-8; PMID:11929805
- Brito DA, Yang Z, Rieder CL. Microtubules do not promote mitotic slippage when the spindle assembly checkpoint cannot be satisfied. J Cell Biol 2008; 182:623-9; PMID:18710927; http://dx.doi.org/10.1083/jcb.200805072
- Yang Z, Kenny AE, Brito DA, Rieder CL. Cells satisfy the mitotic checkpoint in Taxol, and do so faster in concentrations that stabilize syntelic attachments. J Cell Biol 2009; 186:675-84; PMID:19720871; http://dx.doi.org/10.1083/jcb.200906150
- Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol 2015; 16:473-85; PMID:26204159; http://dx.doi.org/10.1038/nrm4025
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer 2010; 10:194-204; PMID:20147901; http://dx.doi.org/10.1038/nrc2803
- Watanabe A, Yasuhira S, Inoue T, Kasai S, Shibazaki M, Takahashi K, Akasaka T, Masuda T, Maesawa C. BCL2 and BCLxL are key determinants of resistance to antitubulin chemotherapeutics in melanoma cells. Exp Dermatol 2013; 22:518-23; PMID:23802633; http://dx.doi.org/10.1111/exd.12185
- Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res 1999; 59:1391-9; PMID:11512523; http://dx.doi.org/10.1054/drup.2001.0193
- Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001; 4:303-13; PMID:11991684; http://dx.doi.org/10.1054/drup.2001.0213
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990; 63:1129-36; PMID:2175676
- Hoppe-Seyler F, Butz K. Repression of endogenous p53 transactivation function in HeLa cervical carcinoma cells by human papillomavirus type 16 E6, human mdm-2, and mutant p53. J Virol 1993; 67:3111-7; PMID:8388491
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Meth 2012; 9:676-82; PMID:22743772; http://dx.doi.org/10.1038/nmeth.2019
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008; 132:487-98; PMID:18267078; http://dx.doi.org/10.1016/j.cell.2007.12.033
- Sakaue-Sawano A, Kobayashi T, Ohtawa K, Miyawaki A. Drug-induced cell cycle modulation leading to cell-cycle arrest, nuclear mis-segregation, or endoreplication. BMC Cell Biol 2011; 12:2; PMID:21226962; http://dx.doi.org/10.1186/1471-2121-12-2
- Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, Sorger PK. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell 2008; 30:11-25; PMID:18406323; http://dx.doi.org/10.1016/j.molcel.2008.02.012
- Weaver BAA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005; 8:7-12; PMID:16023594; http://dx.doi.org/10.1016/j.ccr.2005.06.011
- Demidenko ZN, Kalurupalle S, Hanko C, Lim C-U, Broude E, Blagosklonny MV. Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene 2008; 27:4402-10; PMID:18469851; http://dx.doi.org/10.1038/onc.2008.82
- Cassimeris L, Morabito J. TOGp, the human homolog of XMAP215/Dis1, is required for centrosome integrity, spindle pole organization, and bipolar spindle assembly. Mol Biol Cell 2004; 15:1580-90; PMID:14718566; http://dx.doi.org/10.1091/mbc.E03-07-0544
- Skoufias DA, DeBonis S, Saoudi Y, Lebeau L, Crevel I, Cross R, Wade RH, Hackney D, Kozielski F. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J Biol Chem 2006; 281:17559-69; PMID:16507573; http://dx.doi.org/10.1074/jbc.M511735200
- Torres K, Horwitz SB. Mechanisms of Taxol-induced Cell Death Are Concentration Dependent. Cancer Res 1998; 58:3620-6; PMID:9721870
- Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene 2001; 20:3806-13; PMID:11439344; http://dx.doi.org/10.1038/sj.onc.1204487
- Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell 2004; 7:637-51; PMID:15525526; http://dx.doi.org/10.1016/j.devcel.2004.09.002