
The recent advances in genome-engineering by technologies such as CRISPR/Cas9 now enable an unprecedented level of genome manipulation to support the analysis of phenotypes arising from gene disruption. An alternative approach of gene ablation is the depletion of the gene product by RNA interference (RNAi). While both approaches decrease the level of the protein of interest and so should, at first glance, give similar results, phenotypes caused by disruption of gene function using RNAi and CRISPR/Cas9 can show remarkably little correlation.Citation1 Such phenotypic differences cannot arise solely from off target effects in one of the 2 methods.Citation1 Instead, prolonged gene loss can result in genetic compensation as the system adapts to the long-term consequences of chronic depletion of the target protein. Our analysis of the function of a guanine nucleotide exchange factor (GEF) named Dedicator Of Cytokinesis 6 (DOCK6), has uncovered a striking example of such compensation.
To gain more insights into the function of DOCK6, we used chronic depletion (48 h) of DOCK6 and realized that this led to severe phenotypes impacting upon cell spreading and actin polymerization in interphase cells. However, a longer period of depletion (120 h ≤) resulted in a much milder spreading and actin defects. Moreover the phenotype of DOCK6 knockout (KO) cells emulated those of long term RNAi in having only a relatively mild actin organization defect (). Similarly, the mitotic spindle defects of short term DOCK6 depletion were not observed in DOCK6 KO HeLa cells. After eliminating a possible off target effect of the RNAi with rescue experiments, we hypothesized that a time-scale dependent adaptation mechanism induced by DOCK6 gene disruption compensated for the initial important requirement of this GEF for actin organization. Interestingly, homozygous loss of function mutations in DOCK6 were found to be responsible for Adams-Oliver Syndrome (AOS). AOS manifests itself with skin, scalp, cranium and transverse limb defects.Citation2 Fibroblast cells isolated from AOS patients indicated only minor actin polymerization and spreading defects compared to cells from control individuals. These relative mild phenotypes are again consistent with our hypothesis of DOCK6 gene disruption induced adaptation.Citation2
Figure 1. During early cell spreading DOCK6 dependent RAC1 activation is vital for the cell to balance the activities of RAC1 and RHOA. During acute depletion, RAC1 cannot be activated resulting in a misbalance of RAC1/RHOA activities. Lack of CDC42 activation interferes with the proper mitotic progression. Chronic ablation of DOCK6 is compensated by adaptation that restored CDC42, RAC1 and RHOA activities.
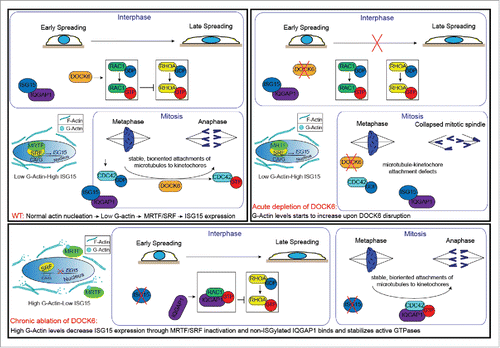
On what level do cells compensate the long-term loss of DOCK6? Chronic depletion of DOCK6 affected the balance between RAC1/RHOA activities which is vital for the initial cell spreadingCitation3 (). In mitosis, however, it was the reduced activity of CDC42 that caused the kinetochore-microtubule attachment defect of short-term DOCK6 siRNA depletion. Interestingly, this RAC1/RHOA misbalance and the reduction in mitotic CDC42 activity were both restored in DOCK6 KO cells. Thus, adaption in interphase and mitosis works by restoring RHO GTPase levels ().
To address the factor(s) that are responsible for the compensation for the loss of DOCK6 function, we conducted an expression profiling experiment with the idea that up- or downregulation of a gene was part of the suppression mechanism. This screen indicated that the expression of Interferon-stimulated gene 15 (ISG15) was lower in DOCK6 KO cells and in AOS patient fibroblasts than in controls. After confirming the downregulation of ISG15 on the protein level, we tested whether reduced ISG15 expression was responsible for the adaptation to DOCK6 function loss. Overexpression of ISG15 or induction of its expression by interferon-α treatment in DOCK6 KO HeLa cells caused a spreading defect reminiscent of those invoked by acute DOCK6 depletion.Citation3 In addition, RNAi-mediated co-depletion of ISG15 together with DOCK6 was able to accelerate adaptation of chronic DOCK6 depletion phenotypes indicating that ISG15 is a key player in the compensation pathway of DOCK6 disruption.Citation3
As for the previously reported mouse homolog, human ISG15 was also under control of myocardin-related transcription factor A/Serum response factor (MRTF-A/SRF) transcription machinery which is sensitive to the levels of monomeric actin (G-actin).Citation5,6 Acute loss of DOCK6 GEF activity led to an increase in the cytoplasmic pool of G-Actin, which then bound and sequestered MRTF-A in the cytoplasm. Decreased levels of nuclear MRTF-A down regulated the transcription of ISG15. This feedback on ISG15 expression explains how cells sense the absence of DOCK6 and respond accordingly in a time-dependent manner.Citation3
ISG15 is a small ubiquitin-like protein modifier that is conjugated to target proteins (also known as ISGylation) through E1, E2 and E3 enzymes similar to ubiquitin. Depletion of the ISG15 E2 enzyme together with DOCK6 was able to accelerate the adaptation just as ISG15 depletion did, indicating that a conjugated form of ISG15 but not the free ISG15 was responsible for the genetic compensation of DOCK6 disruption. We identified the IQ motif containing GTPase activating protein 1 (IQGAP1) as the major target of ISGylation in this case.Citation3 IQGAP1 stabilized the active forms of RAC1 and CDC42 GTPases. IQGAP1 binding has the ability to upregulate the active, GTP bound forms of RAC1 and CDC42.Citation7 For this reason, we tested how ISG15 affected the IQGAP1-RAC1/CDC42 interaction. The affinity of IQGAP1 for active RAC1/CDC42 was higher in ISG15 KO cells than in wild type controls, showing that ISG15 negatively regulates IQGAP1’s affinity toward active RAC1/CDC42.Citation3 Moreover, ISGylation of IQGAP1 was reduced in DOCK6 KO cells. This suggests that ISGylation controls the ability of IQGAP1 to balance the activities of CDC42 and RAC1 in response to perturbation ().
The compensation mechanism described by Cerikan et al. (2016) sheds light on a cellular signaling function of ISG15 that is different to the well-studied role of ISG15 in innate cell immunity. Molecular compensation of DOCK6 disruption through ISGylation opens new horizons in the newly emerging field of ISG15 dependent actin cytoskeleton dynamics.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Morgens DW, Deans R, Li A, Bassik MC. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat Biotechnol 2016; 34:634-636; http://dx.doi.org/10.1038/nbt.3567
- Shaheen R, Faqeih E, Sunker A, Morsy H, Al-Sheddi T, Shamseldin HE, Adly N, Hashem M, Alkuraya FS. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am J Hum Genet 2011; 89:328–33; http://dx.doi.org/10.1016/j.ajhg.2011.07.009
- Cerikan B, Shaheen R, Colo GP, Gläßer C, Hata S, Knobeloch KP, Alkuraya FS, Fässler R, Schiebel E. Cell-intrinsic adaptation arising from chronic ablation of a key rho GTPase regulator. Dev Cell 2016; 39:28-43; PMID: 27693507; http://dx.doi.org/10.1016/j.devcel.2016.08.020
- Chircop M. Rho GTPases as regulators of mitosis and cytokinesis in mammalian cells. Small GTPases 2014; 5:e29770; PMCID: PMC4160341; http://dx.doi.org/10.4161/sgtp.29770
- Hermann M-R, Jakobsen M, Colo GP, Rognoni E, Jakobson M, Kupatt C, Posern G, Fässler R. Integrins synergise to induce expression of the MRTF-A-SRF target gene ISG15 for promoting cancer cell invasion. J Cell Sci 2016; 129:1391–403; PMID: 26872785; http://dx.doi.org/10.1242/jcs.177592
- Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 2010; 11:353–65; http://dx.doi.org/10.1038/nrm2890
- Brown MD, Sacks DB. IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol 2006; 16:242–9; http://dx.doi.org/10.1016/j.tcb.2006.03.002