
ABSTRACT
Our recent study showed that quiescent G0 cells are more resistant to ionizing radiation than G1 cells; however, the underlying mechanism for this increased radioresistance is unknown. Based on the relatively lower DNA damage induced in G0 cells, we hypothesize that these cells are exposed to less oxidative stress during exposure. As a catalytic subunit of NADPH oxidase, Ras-related C3 botulinum toxin substrate 2 (RAC2) may be involved in the cellular response to ionizing radiation. Here, we show that RAC2 was expressed at low levels in G0 cells but increased substantially in G1 cells. Relative to G1 cells, the total antioxidant capacity in G0 phase cells increased upon exposure to X-ray radiation, whereas the intracellular concentration of ROS and malondialdehyde increased only slightly. The induction of DNA single- and double-stranded breaks in G1 cells by X-ray radiation was inhibited by knockdown of RAC2. P38 MAPK interaction with RAC2 resulted in a decrease of functional RAC2. Increased phosphorylation of P38 MAPK in G0 cells also increased cellular radioresistance; however, excessive production of ROS caused P38 MAPK dephosphorylation. P38 MAPK, phosphorylated P38 MAPK, and RAC2 regulated in mutual feedback and negative feedback regulatory pathways, resulting in the radioresistance of G0 cells.
Introduction
Risk assessment of ionizing radiation is crucial to both manned space exploration and radiation protection to normal tissues in radiotherapy. The majority of cells in the human body remain in the G0 phase of the cell cycle. They exist as quiescent or non-cycling cells, and may remain at this stage for days or even years until the resumption of cell division. For example, muscle and nerve cells are permanently arrested in the G0 phase, whereas other cell types such as liver cells resume the cell cycle and enter the G1 phase in response to exogenous stimuli such as injury.Citation1 In a recent study, we showed that G0 cells were more resistant to ionizing radiation when compare with either G1 or exponentially growing cells.Citation2 Earlier studies reported that G0 cells possessed better repair systems than cycling cells.Citation3 However, the underlying mechanism of radioresistance is still unknown.
Ionizing radiation induces biological damages mainly by inducing reactive oxygen species (ROS), a class of highly reactive molecules. Intracellular accumulation of ROS results in gene mutations, carcinogenesis, and ultimately cell death.Citation4-7 Notably, G0 cells exhibit relatively little DNA damages upon exposure to ionizing radiation,Citation2 and based upon this property, we hypothesize that G0 cells possess a biochemical mechanism that prevents accumulation of damaging ROS following ionizing radiation.
The respiratory chain is a source of ROS, and an impaired electron transport chain or mitochondrial DNA damage results in ROS generation.Citation8,9 Ras-related C3 botulinum toxin substrate 2 (RAC2) is a GTPase of ∼21 kDa containing the catalytic subunit of NADPH oxidase, which is the main component of the respiratory chain.Citation10,11 GTPases are crucial regulators of diverse cell processes including cell growth, cytoskeletal reorganization, and activation of protein kinases.Citation12-15 Several studies have reported that RAC2 plays an essential role in the regulation of superoxide production in neutrophils.Citation16 However, its role in the regulation of cellular responses to ionizing radiation is less known.
P38 MAPK is a 38 kDa protein,Citation17 which may be activated by multiple exogenous stimuli such as ultraviolet radiation, cytotoxic chemicals and ionizing radiation.Citation18 P38 MAPK activation regulates gene expression and downstream cytokines. It plays an important role in the regulation of the cell cycle checkpoint, arresting cells in the G1/S transition, G2/M checkpoint, and inhibiting cell proliferation by downregulating cyclin D1. All of these biochemical processes may affect the radiosensitivity of cells.
Nieborowska-Skorska et al. reported that Rac2 generated high levels of ROS promote oxidative DNA damage to trigger genomic instability in chronic myeloid leukemia stem cells.Citation19 Therefore, we proposed the hypothesis that RAC2 as well as P38 MAPK might play an important role in the G0 cells radioresistance. In order to characterize the mechanisms responsible for the resistance of G0 cells, also by bioinformatics prediction, we therefore identified the regulatory functions of RAC2 and P38 MAPK in the generation of ROS and DNA damages after exposure to ionizing radiation.
Results
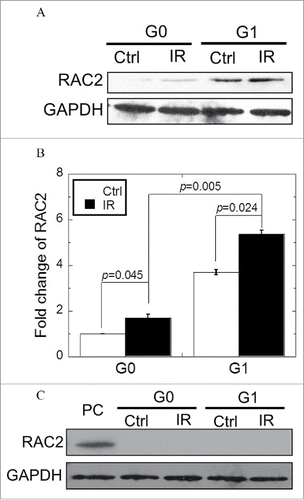
Differential expression of RAC2 protein was observed in G0 and G1 cells
In order to investigate RAC2 expression in G0 and G1 cells, we performed western blot analyses and found that prior to irradiation, extremely small quantities of RAC2 expression were observed in G0 cells, whereas strong signals for RAC2 protein were detected for cell lysates prepared from G1 cells (). When G0 cells were exposed to 2 Gy X-ray radiation, RAC2 expression levels were only slightly upregulated (p = 0.045), while G1 cells showed greater upregulation (p = 0.024). In order to decrease RAC2 expression to the same level in the 2 cell types, RNAi technology was used. Western blotting confirmed that RAC2 shRNA treatment resulted in efficient inhibition of RAC2 protein expression, and no detectable RAC2 was observed in either G0 or G1 cells independent of exposure to 2 Gy X-ray radiation ().
Figure 1. Expression of RAC2 in G0 and G1 cells. (A) The expression of RAC2 was measured by western blotting. (B) Grayscale analysis of RAC2 in G0 and G1 cells. (C) RNAi was used to silence RAC2 expression and western blotting was used to check the inhibition efficiency of shRNA. PC: positive control. Error bars denote mean ± SE derived from 3 independent experiments.
Higher NADPH oxidase activity was decreased by the knockdown of RAC2, which enhanced the generation of ROS
In order to determine the dependence of NADPH oxidase activity (short as NOA below) on RAC2 levels, the NOA was measured using a commercial kit. The reaction, and, therefore, the NOA in G0 cells was lower than that for G1 cells (p = 0.044 in ). Although both G0 and G1 cells showed an increase in NOA following exposure to 2 Gy X-ray radiation, G1 cells had a greater reaction (p = 0.037) when background NOA were subtracted (). Upon addition of the NADPH oxidase specific inhibitor, diphenyleneiodinium (DPI), the NOA was inhibited in both cell types, independent of radiation. Consistent with these results, the absence of RAC2 resulted in a reduction in the reaction of NADPH in both G0 and G1 cells, suggesting that RAC2 plays a major role in determining the cellular NOA (). Because RAC2 serves as an important activating subunit of NADPH oxidase, we hypothesized that RAC2 is involved in the generation of ROS. We conducted a DCF-DA assay to detect the levels of intracellular ROS and found that the levels of ROS increased dramatically in G1 cells, whereas in G0 cells there was little change after exposure to 2 Gy of X-ray radiation (p = 0.008 in G0 cells and p = 0.003 in G1 cells) ( and ). Upon 2 Gy X-ray radiation, the levels of ROS increased only slightly in RAC2-knockdown G1 cells when compared with wild-type G1 cells (p = 0.007). As expected, low levels of ROS remained in G0 cells regardless of the RAC2 expression level.
Figure 2. NADPH consumption and oxidative stress of G0 and G1 cells after exposure. (A) G0 and G1 cells transfected with control shRNA (NC shRNA). (B) G0 and G1 cells transfected with RAC2 shRNA. Diphenyleneiodinium (DPI) (5 μM) was used to inhibit the activity of NADPH oxidase. (C, D). The concentration of ROS in G0 and G1 cells irradiated by 2 Gy X-raywas measured by a DCF-DA assay. Images were captured at an exposure time of 0.035 seconds and the intensity of the dichlorofluorescein (DCF) fluorescence was quantified using ImageJ software. (E) Total antioxidant capacity (TAC) and (F) Malondialdehyde (MDA) yields were measured in G0 and G1 cells of different treatments. Error bars denote the mean ± SE derived from 3 independent experiments. NC shRNA, the negative control of shRNA.
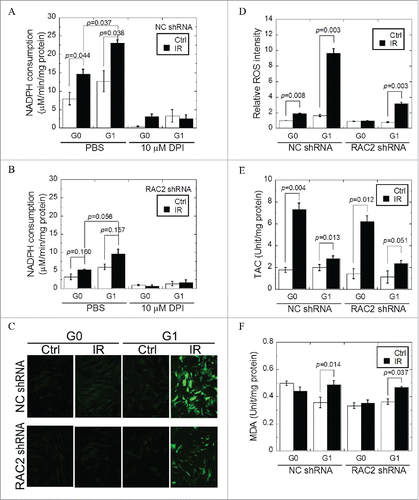
The low levels of ROS measured in G0 cells suggested that these cells have a higher antioxidant capacity. In order to confirm this, we compared both total antioxidant capacity (TAC) and malondialdehyde (MDA) yields between G0 and G1 cells. As shown in following exposure to 2 Gy X-ray radiation, the TAC of G0 cells increased more than that of G1 cells (p = 0.028). In addition, X-ray radiation did not increase the concentration of MDA, a product of lipid peroxidation in G0 cells, but in G1 cells (p = 0.014) (). Moreover, knockdown of RAC2 had no significant effect on the concentration of MDA in either G0 or G1 cells. Taken together, these results suggest that RAC2 does not play an important role in the resistance of G0 cells to oxidative stress.
Low RAC2 expression results in increased cellular resistant to ionizing radiation
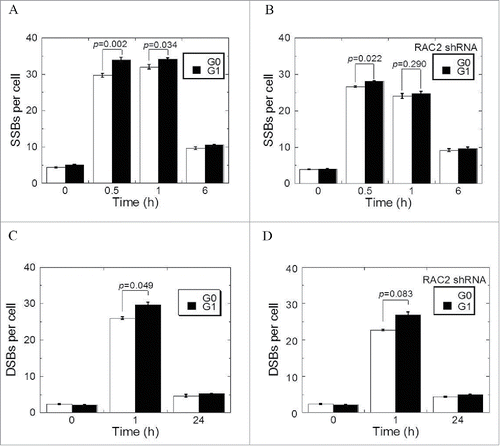
Excessive accumulation of ROS may result in the induction of DNA single-strand breaks (SSBs). Thus, we compared the induction of DNA SSBs as shown by XRCC1 foci in G0 cells to that in G1 cells following 2 Gy X-ray radiation. Radiation induced more XRCC1 foci in G1 cells than in G0 cells, as measured at 30 minutes (p = 0.002) and 1 hour (p = 0.034) after irradiation (). XRCC1 foci reduced from 31.99 ± 0.62(1h) to 9.67 ± 0.35 (6h) in G0 and 34.10 ± 0.53 0.32 (1h) to 10.52 ± 0.11 (6h) in G1 cells. The remaining XRCC1 foci were not significantly affected by the cell cycle phase. Knocking down of RAC2 reduced the induction of XRCC1 foci in both G1 as well as in G0 cells. However, the reduction in G1 cells was higher such the difference in SSB induction between G1 and G0 cells diminished (). To confirm these observations, we measured both induction and repair of DNA-double stranded breaks (DSBs) with 53BP1 foci. Very similar results to DNA SSBs were obtained ( and ), with the induction of DNA damage in G1 cells more reduced in the absence of RAC2.
Figure 3. The kinetics of DNA damage and repair. (A, B) XRCC1 foci induced by 2 Gy X-ray radiation of G0 and G1 cells transfected with control shRNA (A) or RAC2 shRNA (B). (C, D) The 53BP1 foci induced by 2 Gy X-ray irradiation of G0 and G1 cells transfected with control shRNA (C) or RAC2 shRNA (D). Error bars denote the mean ± SE derived from 3 independent experiments.
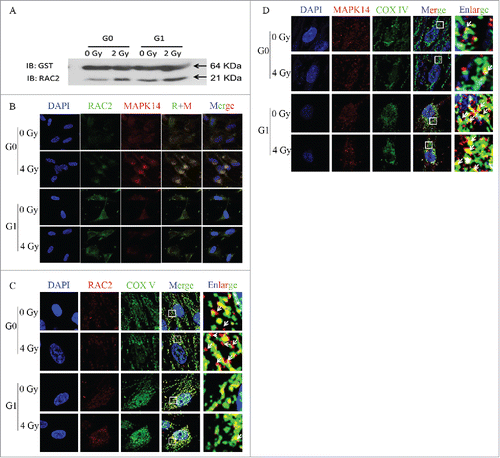
RAC2 and P38 MAPK are co-localized and assembled in G0 cells
A GST pull down assay was used to characterize the relationship between RAC2 and P38 MAPK. The GST-tagged α-subunit of P38 MAPK (MAPK14) was transfected into cells. The results showed that RAC2 could be pulled down by the MAPK14-GST fusion protein (). The intracellular localization was determined using an immunofluorescence assay. MAPK14 and RAC2 were intracellularly co-localized prior to and after exposure (). When P38 MAPK or RAC2 was hybridized with mitochondria using fluorescent labeled cytochrome C-oxidase (COX IV) as a probe. All the RAC2 co-localized with mitochondria in G1 cells with or without irradiation, but in G0 cells, some RAC2 did not co-localize with mitochondria (). However, P38 MAPK was found to be highly co-localized with mitochondria, but did not completely overlap both in G0 and G1 cells, with or without irradiation (). Taken together, the results showed that RAC2 that was not co-localized with mitochondria could not catalyze the production of ROS in G0 cells.
Figure 4. RAC2 and P38 MAPK co-localize and assemble in G0 cells after exposure. (A) A GST pull down assay shows the interaction of P38 MAPK and RAC2. (B, C, and D), and the intracellular protein hybridization of P38 MAPK and RAC2. (B) RAC2 and P38 MAPK are co-localized in cells. (C) RAC2 and mitochondria co-localization. (D) P38 MAPK and mitochondria co- localization. COX IV was used as a mitochondrial marker.
Phosphorylation of P38 MAPK increased cellular radioresistance
Prior to irradiation, large quantities of phosphorylated P38 MAPK expression were found in both G0 and G1 cells ( and ). After exposure to a 2 Gy dose of X-rays, phosphorylated P38 MAPK was slightly upregulated in G0 cells, whereas the level decreased in G1 cells (p = 0.028) ( and ). To characterize the relationship between phosphorylated P38 MAPK and cellular radiosensitivity, we used SB203580, a selective inhibitor of phosphorylated P38 MAPK, to reduce the phosphorylated P38 MAPK levels in G0 cells to that of G1 cells. Thus, we compared the survival of G0 cells in the absence or presence of 20 µM SB203580 with the survival of G1 cells with or without a 2 Gy dose of X-rays irradiation. The percentage survival was unchanged when G0 cells were incubated with SB203580, while there was lower survival in SB203580 and radiation-treated G0 cells compared with the control (p = 0.027). The percentage survival was even lower than G1 cells (p = 0.009) after exposure (). Micronuclei frequency in SB203580-treated G0 cells was also higher than the negative G0 cells (p = 0.004) (). After exposure, when the G0 cells were treated with SB203580, the expression levels of Bcl−2 were downregulated while Bax were upregulated (). The levels of Bcl−2 and Bax promoted apoptosis when phosphorylated P38 MAPK was inhibited ().
Figure 5. Phosphorylated P38 MAPK increases the radioresistance of G0 cells. (A) The expression of P38 MAPK. (B) The expression of phosphorylated P38 MAPK. (C, D) Grayscale analyses of P38 MAPK (C) and phosphorylated P38 MAPK (D). (E) Survival of cells treated with IR with or without SB203580. (F) The amount of micronuclei in cells treated with IR with or without SB203580. (G) Bcl−2 and Bax expression when the cells were treated with IR with or without SB203580. (H) Grayscale analysis of Bcl−2 and Bax when the cells were treated with IR with or without SB203580. Error bars denote the mean ± SE derived from 3 independent experiments.
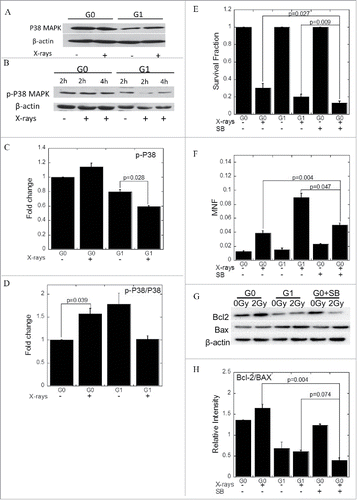
ROS induced dephosphorylation of P38 MAPK
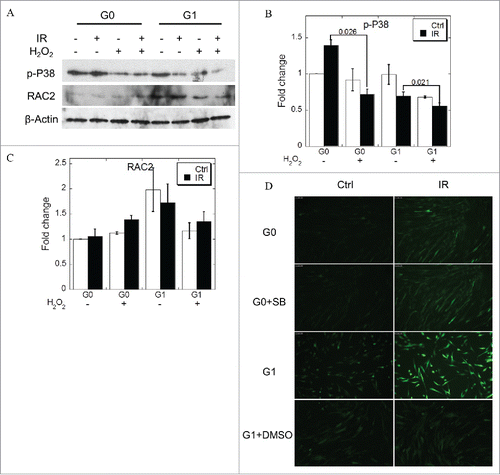
We then characterized the relationship between ROS and phosphorylated P38 MAPK levels in the MRC-5 cells. We increased the levels of ROS in the medium using hydrogen peroxide to further ascertain the level of phosphorylated P38 MAPK in cells under oxidative stress. As shown in , ROS had no effect on RAC2 expression ( & ), while caused the dephosphorylation of P38 MAPK ( & ). As shown in , we treated G1 cells with DMSO and confirmed the reduction of ROS concentration. While when G0 cells were treated with SB203580, no difference in the yields of ROS from control group was observed. In addition, the phosphorylated P38 MAPK did not affect the generation of ROS ().
Figure 6. ROS induces dephosphorylation of P38 MAPK. (A) Phosphorylated P38 MAPK and RAC expression when the cells were treated with excess H2O2. (B, C) Grayscale analyses of P38 MAPK (B) and RAC2 (C). (D) ROS concentration when the cells were treated with IR with or without SB230580. DMSO was used as positive control. Error bars denote the mean ± SE derived from 3 independent experiments.
Discussion
In the present study, we showed that low expression of RAC2 contributed to the relatively high resistance of G0 cells to ionizing radiation, resulting from the low NOA. This led to high levels of NADPH but low levels of intracellular ROS, resulting in minimal DNA damage. These findings were confirmed in G1 cells, which possess the same genetic background and the same DNA content as G0 cells, but higher RAC2 protein levels and higher radiosensitivity ().
Table 1. Different responses of G0 and G1 cells to ionizing radiation.
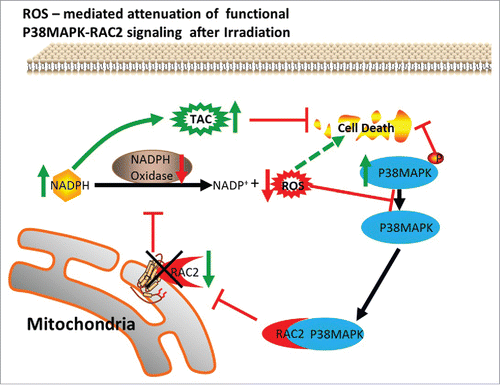
The ROS, RAC2, and P38 MAPK feedback loop causes G0 cells to become more resistant to ionizing radiation. 1) After irradiation, the expression of RAC2 in G0 cells was lower than in G1 cells, resulting in a lower NOA, thereby reducing the concentration of ROS in G0 cells. G0 cells avoided ROS-induced DSBs and SSBs. In addition, the low NOA led to a decreased reaction of NADPH in G0 cells. High concentrations of NADPH promoted the reduction of glutathione peroxidase from an oxidized state to a reduced state. The TAC in G0 cells was higher than that in G1 cells. 2) The α subunit of P38 MAPK interacted with RAC2, resulting in RAC2 that was not localized to the mitochondria. The decrease of functional RAC2 leads to low levels of ROS in G0 cells. 3) Phosphorylated P38 MAPK was increased in G0 cells while reduced in G1 cells after irradiation. Phosphorylated P38 MAPK increased cellular radioresistance. 4) Excessive production of ROS caused cellular P38 MAPK dephosphorylation. 5) P38 MAPK, phosphorylated P38 MAPK, and RAC2 regulated a mutual feedback and negative feedback regulatory pathway, leading to the radioresistance of G0 cells ().
Figure 7. The ROS, RAC2, and P38 MAPK feedback loop induces resistance of G0 cells to ionizing radiation.
The low expression of RAC2 accounts for the cellular radioresistance by suppressing ROS production and reducing the reaction of NADPH, which results in high antioxidant activity that further suppresses the generation of ROS. ROS is produced not only by the interaction of ionizing radiation with water and bio-molecules in cells, but also by intracellular biological processes. NADPH oxidase catalyzes the production of superoxide from oxygen and NADPH. It is a major source of intracellular ROS in mammalian cells and modulates the activation of redox-sensitive signaling pathways that influence the intracellular environment, adhesion molecule expression, permeability, growth, migration, and cellular functions.Citation20-22 Quiescent fibroblasts exhibit high activity of the pentose phosphate pathway,Citation21 which does not generate ATP but forms NADPH.Citation24 Reduced glutathione (GSH), NADPH-producing pathways, and glutathione reductase play important roles in intracellular redox homeostasis.Citation25 NADPH-dependent glutathione reductase is a member of the FAD-binding disulfide reductase superfamily and the major enzyme responsible for reduction of GSSG in most organisms, which provides a defense system against oxidants.Citation26-28 GSH oxidation also causes stimulation of the hexose monophosphate shunt and increases the production of NADPH.Citation26 The low consumption of NADPH and low yields of ROS are associated with low expression levels of RACs in G0 cells, which result in a high antioxidant capacity and radioresistance.
DPI is a specific inhibitor of NADPH oxidase, which completely inhibited the activity of NADPH oxidase. Compared to X-ray exposure, knockdown of RAC2 resulted in an incomplete reduction in the reaction of NADPH in both G0 and G1 cells, suggesting that RAC2 plays a major but not exclusive role in determining the activity of NADPH oxidase. Other processes might also be involved in the reaction of NADPH. For example, the reduction of GSSG by glutathione reductase also involves NADPH.Citation26
ROS damages DNA bases or the sugar backbone that lead to SSBs.Citation29 DSBs also result from ROS-induced lesionsCitation30; however, indirect interactions mediated by ROS dominates during X-ray exposure, so yields of SSBs are higher than DSBs. This may explain why SSBs, and not DSBs, predominantly decrease in response to the reduction of ROS when RAC2 is knocked down with shRNA.
In the present study, we characterized the relationship of RAC expression levels to cellular radiosensitivity. Low expression of RAC2 leads to a low reaction of NADPH and low yields of ROS. A reduction in ROS production in G0 cells, together with increased levels of ROS scavengers, provide a high antioxidant capacity but low susceptibility to ionizing radiation. These findings provide new insights into the radioresistance of G0 cells, and suggest a potential application for RAC2 inhibitors as radioprotectors.
Materials and methods
Cell culture and irradiation
Human embryonic lung fibroblast cell line MRC-5 Citation31 was maintained in MEM (Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA), 1% penicillin sodium and 1% streptomycin, at 37°C. G0 cells and G1 cells were obtained as previously described.Citation30 In brief, 5 × 105 cells plated in T25 flasks were allowed to grow for 8 d to become quiescent (G0 phase) by contact inhibition with one medium refreshment on day 4. Then, cells were harvested, subcultured in a new flask of the same size and incubated for 6 h in fresh medium to allow them to reach G1 phase. X-ray irradiation was implemented with the X-ray generator (Faxitron 650, Faxitron Bioptics, Lincolnshire, IL, USA), which was operated with 100 kVp and 5 mA at room temperature.
Gene silencing with shRNA
Cells were seeded in 12-well plates (1 × 105 cells per well) with antibiotic-free MEM medium containing 10% serum 48 h before transfection. Cells were transfected with 1 × 106 RAC2 shRNA lenti-virus particles (Invitrogen, Eugene, OR, USA) for 24 h. Control shRNA and GFP lentivirus particles were also operated according to the manufacturer's instruction. Cells were subsequently cultured with MEM medium contains 1 μM puromycin (Invitrogen). Cells stably expressing RAC2 siRNA double-checked with Western blot were used for further experiments.
Immunofluorescence
Irradiated cells were fixed for 10 min in 4% paraformaldehyde, permeabilized for 20 min in methanol at −20 °C, blocked for 1 h with 5% skim milk, and stained with mouse anti-53BP1 antibody or XCRR1, P38 MAPK and RAC2 (Upstate Biotechnology, Lake Placid, NY, USA) for 2 h. The bound antibody was visualized using Alexa Fluor® 594 anti-mouse antibody (Molecular Probes, Eugene, OR, USA) and cell nuclei were counterstained with DAPI (PharMingen, San Jose, CA, USA). Slides were observed under a confocal laser scanning microscope (Nikon, Tokyo, Japan). At least 100 cells were scored for each sample and the average number of foci per cell was calculated.
Western blot
Total proteins were separated by 12% SDS-PAGE and transferred to hybond nitrocellulose membrane (Amersham, NJ, USA). The membrane was blocked with 5% nonfat milk powder in Tris-buffered saline (pH 7.5) and hybridized overnight with primary antibodies (P38 MAPK, phosphorylated P38 MAPK, RAC2, COX IV, Actin), which were then detected with horseradish peroxidase-conjugated anti-IgG and visualized with an ECL kit (Amersham). Our proteins were normalized to the loading controls.
Measurement of ROS
ROS production was measured using the DCF-DA assay.Citation33,35 In brief, MRC-5 cells were seeded in Ф60 mm culture dishes at 70% confluence and then treated with ionizing radiation. After treatment, cells were incubated with 10 μM of DCF-DA (Sigma) for 10 min. The cells were harvested, washed once, and re-suspended in PBS. Excitation light was monitored using a fluorescence microscope under 485nm. Photos were taken at precision exposure time. The mean of the DCF fluorescence intensity was obtained from different photos. Fluorescence intensity was quantified by software.
Measurements of antioxide parameters
Cells were harvested after irradiation and homogenized. After adjusted to appropriate concentrations, cells were homogenized with liquid nitrogen and divided into several portions. After protein quantification, the activity of superoxide dismutase (SOD), total anti-oxide capacity (TAC) and the yield of lipid peroxidation product, malondialdehyde (MDA), were measured with colorimetric/fluorometric assay kit (Sigma) by following the manufacturer's instructions.
NADPH oxidase activity
NADPH consumption, which reflecting the activity of NADPH oxidase, was measured at 530 nm by using the nitro blue tetrazolium NBT (Beyotime, Nanjing, China) as an electron acceptor with a spectrophotometer (Aminco DW2000, SLM Instruments, Urbana, IL). The reaction mixture consisted of a Tris-buffer (50 mM Tris-HCl, pH 7.4, 250 mM SUC). 100 μM of NADPH was added as the substrate. Its inhibitor diphenyleneiodinium (DPI, 10 μM, Beyotime) was added 5 min before plate-reading. The reaction was started by the addition of 0.1 mL of homogenate.
Statistics
All experiments were independently repeated at least 3 times and all data were presented as the mean ± standard error. Student's t-tests were used for statistic analysis. Probability (p) values less than 0.05 were considered to be statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed
Author Contributions
Hailong Pei and Jian Zhang designed the study and conducted data analysis. Jing Nie managed laboratory work, including quality assurance and control. These authors contributed equally to this work. Nan Ding, Wentao Hu and Junrui Hua provided some help on technical field. Ryoichi Hirayama, Yoshiya Furusawa, Cuihua Liu provide a lot of help during the radiation exposure at the Quantum and Radiological Science and Technology, Japan. Bingyan Li, Tom K. Hei provided valuable suggestion and revised the English writing. Guangming Zhou designed the study and organized the study and the whole research work.
Funding
This work was supported by the National Natural Science Foundations of China awarded (No. 11335011, 31370846, 31400723, 11405235, 31600674 and 81602794), China Postdoctoral Science Foundation funded project (2016M591904). Natural Science Foundations of Jiangsu awarded (BK20160334).
References
- Ginn-Pease ME, Eng C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Res 2003; 63:282-6; PMID:12543774
- Ding N, Pei H, He J, Furusawa Y, Hirayama R, Liu C, Matsumoto R, Li H, Hu W, Li Y, et al. Simulated studies on the biological effects of space radiation on quiescent human fibroblasts. Adv Space Res 2013; 52:1314-9; http://dx.doi.org/10.1016/j.asr.2013.06.030
- Elsasser T, Weyrather WK, Friedrich T, Durante M, Iancu G, Krämer M, Kragl G, Brons S, Winter M, Weber KJ, et al. Quantification of the relative biological effectiveness for ion beam radiotherapy: direct experimental comparison of proton and carbon ion beams and a novel approach for treatment planning. Int J Radiat Oncol Biol Phys 2010; 78:1177-83; PMID:20732758; http://dx.doi.org/10.1016/j.ijrobp.2010.05.014
- Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD. Free radicals and antioxidants in human health: current status and future prospects. J Assoc Physicians India 2004; 52:794-804; PMID:15909857
- Chae HJ, Chae SW, Kang JS, Bang BG, Han JI, Moon SR, Park RK, So HS, Jee KS, Kim HM, et al. Effect of ionizing radiation on the differentiation of ROS 17/2.8 osteoblasts through free radicals. J Radiat Res 1999; 40:323-35; PMID:10748578; http://dx.doi.org/10.1269/jrr.40.323
- Du C, Gao Z, Venkatesha VA, Kalen AL, Chaudhuri L, Spitz DR, Cullen JJ, Oberley LW, Goswami PC. Mitochondrial ROS and radiation induced transformation in mouse embryonic fibroblasts. Cancer Biol Ther 2009; 8:1962-71; PMID:19738419; http://dx.doi.org/10.4161/cbt.8.20.9648
- Jiang Q, Zhou C, Healey S, Chu W, Kouttab N, Bi Z, Wan Y. UV radiation down-regulates Dsg-2 via Rac/NADPH oxidase-mediated generation of ROS in human lens epithelial cells. Int J Mol Med 2006; 18:381-7; PMID:16820949
- Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T, Majima HJ. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007; 7:106-18; PMID:17307400; http://dx.doi.org/10.1016/j.mito.2006.11.026
- Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res 2001; 61:3894-901; PMID:11358802
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol 2006; 16:522-9; PMID:16949823; http://dx.doi.org/10.1016/j.tcb.2006.08.006
- Diebold BA, Bokoch GM. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat Immunol 2001; 2(3):211-5; PMID:11224519; http://dx.doi.org/10.1038/85259
- Fattouh R, Guo CH, Lam GY, Gareau MG, Ngan BY, Glogauer M, Muise AM, Brumell JH. Rac2-deficiency leads to exacerbated and protracted colitis in response to citrobacter rodentium infection. PloS One 2013; 8:e61629; PMID:23613889; http://dx.doi.org/10.1371/journal.pone.0061629
- Shen L, Gao Y, Qian J, Wu Y, Zhou M, Sun A, Zou Y, Ge J. The role of SDF-1alpha/Rac pathway in the regulation of endothelial progenitor cell polarity; homing and expression of Rac1, Rac2 during endothelial repair. Mol Cell Biochem 2012; 365:1-7; PMID:21964561; http://dx.doi.org/10.1007/s11010-011-1083-z
- Shao X, Miao M, Qi X, Chen Z. Ras-proximate-1 GTPase-activating protein and Rac2 may play pivotal roles in the initial development of myelodysplastic syndrome. Oncol Lett 2012; 4:289-98; PMID:22844372; http://dx.doi.org/10.3892/ol.2012.736
- Courjal F, Chuchana P, Theillet C, Fort P. Structure and chromosomal assignment to 22q12 and 17qter of the ras-related Rac2 and Rac3 human genes. Genomics 1997; 44:242-6; PMID:9299243; http://dx.doi.org/10.1006/geno.1997.4871
- Irani K, Goldschmidt-Clermont PJ. Ras, superoxide and signal transduction. Biochem Pharmacol 1998; 55:1339-46; PMID:10076523; http://dx.doi.org/10.1016/S0006-2952(97)00616-3
- Morazzani M, de Carvalho DD, Kovacic H, Smida-Rezgui S, Briand C, Penel C. Monolayer versus aggregate balance in survival process for EGF-induced apoptosis in A431 carcinoma cells: Implication of ROS-P38 mapk-integrin A2B1 pathway. Int J Cancer 2004; 110:788-99; PMID:15170659; http://dx.doi.org/10.1002/ijc.20198
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001; 81:807-69; PMID:11274345
- Nieborowska-Skorska M, Kopinski PK, Ray R, Hoser G, Ngaba D, Flis S, Cramer K, Reddy MM, Koptyra M, Penserga T, et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012; 119(18):4253-63; PMID:22411871; http://dx.doi.org/10.1182/blood-2011-10-385658
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000; 87:840-4; PMID:11073878; http://dx.doi.org/10.1161/01.RES.87.10.840
- Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol 2004; 287:R1014-30; PMID:15475499; http://dx.doi.org/10.1152/ajpregu.00124.2004
- Dworakowski R, Alom-Ruiz SP, Shah AM. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol Rep: PR 2008; 60:21-8
- Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, Pollina EA, Rabitz HA, Rabinowitz JD, Coller HA. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol 2010; 8:e1000514; PMID:21049082; http://dx.doi.org/10.1371/journal.pbio.1000514
- Kruger NJ, von Schaewen A. The oxidative pentose phosphate pathway: structure and organisation. Curr Opin Plant Biol 2003; 6:236-46; PMID:12753973; http://dx.doi.org/10.1016/S1369-5266(03)00039-6
- Scott MD, Zuo L, Lubin BH, Chiu DT. NADPH, not glutathione, status modulates oxidant sensitivity in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Blood 1991; 77:2059-64; PMID:2018843
- Marty L, Siala W, Schwarzländer M, Fricker MD, Wirtz M, Sweetlove LJ, Meyer Y, Meyer AJ, Reichheld JP, Hell R. The NADPH-dependent thioredoxin system constitutes a functional backup for cytosolic glutathione reductase in Arabidopsis. Proc Natl Acad Sci U S A 2009; 106:9109-14; PMID:19451637; http://dx.doi.org/10.1073/pnas.0900206106
- Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Müller HM, Botella-Munoz J, Schneuwly S, Schirmer R, Becker K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science 2001; 291:643-6; PMID:11158675; http://dx.doi.org/10.1126/science.291.5504.643
- Krauth-Siegel RL, Comini MA. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim Biophys Acta 2008; 1780:1236-48; PMID:18395526; http://dx.doi.org/10.1016/j.bbagen.2008.03.006
- Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993; 362:709-15; PMID:8469282; http://dx.doi.org/10.1038/362709a0
- Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res 2011; 39:6986-97; PMID:21596788; http://dx.doi.org/10.1093/nar/gkr331
- Jacobs JP, Jones CM, Baille JP. Characteristics of a human diploid cell designated MRC-5. Nature 1970; 227:168-70; PMID:4316953; http://dx.doi.org/10.1038/227168a0
- Hu W, Pei H, Li H, Ding N, He J, Wang J, Furusawa Y, Hirayama R, Matsumoto Y, Liu C. Effects of shielding on the induction of 53BP1 foci and micronuclei after Fe ion exposures. J Radiat Res 2013; 55:10-6; PMID:23728321; http://dx.doi.org/10.1093/jrr/rrt078
- Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radical Bio Med 1999; 27:612-6; http://dx.doi.org/10.1016/S0891-5849(99)00107-0
- Aruoma OI, Grootveld M, Bahorun T. Free radicals in biology and medicine: from inflammation to biotechnology. Biofactors 2006; 27:1-3; PMID:17012759; http://dx.doi.org/10.1002/biof.5520270101