
ABSTRACT
NADPH oxidase 4 (NOX4) and the NOX4-related redox signaling are implicated in cardiac hypertrophy. NOX4 is interrelated with endoplasmic reticulum stress (ERS). Spliced X-box binding protein 1 (Xbp1s) is a key mediator of ERS while its role in cardiac hypertrophy is still poorly understood. Recently, receptor interacting protein kinase 1(RIPK1) has been increasingly reported to be associated with ERS. Therefore, we aimed to test the hypothesis that Xbp1s mediates NOX4-triggered cardiac hypertrophy via RIPK1 signaling. In the heart tissue of transverse aortic constriction (TAC) rats and in primary cultured neonatal cardiomyocytes(NCMs) treated with angiotensinII(AngII) or isoproterenol (ISO), NOX4 expression and reactive oxygen species (ROS) generation, and expression of Xbp1s as well as RIPK1-related phosphorylation of P65 subunit of NF-κB were elevated. Gene silencing of NOX4 by specific small interfering RNA (siRNA) significantly blocked the upregulation of NOX4, generation of ROS, splicing of Xbp1 and activation of the RIPK1-related NF-κB signaling, meanwhile attenuated cardiomyocyte hypertrophy. In addition, ROS scavenger (N-acetyl-L-cysteine, NAC) and NOX4 inhibitor GKT137831 reduced ROS generation and alleviated activation of Xbp1 and RIPK1-related NF-κB signaling. Furthermore, splicing of Xbp1 was responsible for the increase in RIPK1 expression in AngII or ISO-treated NCMs. Upregulated RIPK1 in turn activates NF-κB signaling in a kinase activity-independent manner. These findings suggest that Xbp1s plays an important role in NOX4-triggered cardiomyocyte hypertrophy via activating its downstream effector RIPK1, which may prove significant for the development of future therapeutic strategies.
Introduction
Cardiac hypertrophy, an independent risk factor of cardiac morbidity and mortality, results from the cardiac overload, hyperactivated neurohumoral systems and noxious metabolites.Citation1 Cardiomyocyte hypertrophy is a complex cellular reprogramming process involving myriads of biomolecular mechanisms, among which endoplasmic reticulum stress (ERS) and reactive oxygen species (ROS) are increasingly regarded as important mechanisms of cardiac hypertrophy.Citation2-6
ERS, the disruption of ER homeostasis, initiates 3 major signaling pathways to restore ER homeostasis. These signaling pathways, collectively known as unfolded protein response (UPR), comprise protein kinase RNA-like ER kinase (PERK)/eukaryotic initiation factor 2α(eIF2α)/activating transcriptional factor 4(ATF4), inositol-requiring protein 1α (IRE1α)/Xbp1s and activating transcriptional factor 6 (ATF6) signaling pathways.Citation7 Phosphorylation of IRE1α activates its endoribonuclease activity to splice Xbp1 mRNA, generating spliced Xbp1 (Xbp1s) which is an important component of the UPR and a potent nuclear transcription factor. Over the past decade, a growing number of studies have reported that Xbp1s is involved in a wide range of pathophysiological processes, namely, protein folding, glycosylation, ER-associated degradation, autophagy, lipid biogenesis and insulin secretion.Citation8,9 Of particular note, accumulating evidence suggests that Xbp1s exerts potential regulator effects in cardiovascular diseases, including atherosclerosis, myocardial ischemia/reperfusion injury, and cardiac hypertrophy.Citation8,10 But so far, the precise functions of Xbp1s in cardiomyocyte hypertrophy are still poorly understood.
RIPK1, a signaling hub dictating the cell response to stress,Citation11,12 is increasingly reported to play an important role in cardiac injury induced by ischemia and heart failure.Citation13-15 Importantly, a few studies indicate that IRE1α and Xbp1 interact with RIPK1 to promote cellular survival and growth.Citation16,17 Moreover, our previous work has demonstrated that RIPK1 plays an important role in palmitic acid-induced cardiomyocyte hypertrophy.Citation18 Therefore, these facts raise an intriguing possibility that Xbp1s/RIPK1 may act as a novel signaling pathway in the development of cardiac hypertrophy.
On the other hand, ROS-dependent modification of certain key signaling pathways is involved in the pathogenesis of cardiac hypertrophy.Citation4 Among various intracellular ROS sources, NADPH oxidases(NOXs) is the only professional ROS-generating cellular enzymes.Citation19 NOX4, one of the predominant isoforms in cardiomyocytes, is primarily localized in the membrane of intracellular compartments, such as mitochondria, endoplasmic reticulum(ER) and nucleus.Citation20 As a dedicated ROS generator located in the membrane of ER, activated NOX4 is interrelated with one of the specific ERS signaling pathways under given pathological situation.Citation21-24 Based on aforementioned findings, we postulated that NOX4 and NOX4-derived ROS may promote cardiac hypertrophy through activation of Xbp1s/RIPK1 pathway.
Accordingly, this study was aimed to determine (1) whether activation of Xbp1s is involved in NOX4-triggered cardiac hypertrophy. If so,(2) whether RIPK1 is a downstream effector of Xbp1s in NOX4-induced cardiac hypertrophy.
Results
Upregulation of NOX4 is required for hypertrophic stimuli-induced cardiomyocyte hypertrophy
Firstly, we evaluated the change of NOX4 expression in TAC rats compared with that in sham-operated rats. As shown in , TAC rats showed marked cardiomyocyte hypertrophy with larger LVMI, DPWT, DSVT and cross sectional area. NOX4 protein expression was increased about 4.2-fold in TAC rats compared with sham-operated rats ().
Figure 1. NOX4 expression was increased in cardiac hypertrophy. (A) Echocardiographic analysis for cardiac hypertrophic indicators(DPWT, DVST, LVMI) in rats 4 weeks after TAC or sham surgery. (B) H&E staining for cross sectional area of caridomyocytes in TAC rats and sham-operated rats. (C) Western blotting analysis for NOX4 expression in heart tissue of TAC or sham-operated rats. (D) mRNA expression of NOX4 and hypertrophic markers(ANP, BNP and β-MHC) in NCMs treated with AngII (10−5M for 24h, Top) or ISO(10−5M for 24h, Bottom). (E) Western blotting analysis for NOX4 in NCMs treated with AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom). (F) FCM analysis for ROS generation in NCMs treated with AngII (10−5M for 24h) or ISO (10−5M for 24h). (G) Immunofluorescent staining for NOX4 expression (red) in NCMs treated with AngII (10−5M for 24h) or ISO (10−5M for 24h). (H) Surface area determination for NCMs treated with AngII (10−5M for 24h) or ISO (10−5M for 24h). Error bars indicate SEM. n = 6 for A–C and n = 4 for D-H. The fluorescent micrograph is representative of cells from 4 independent visual fields. * indicates P<0.05 versus control group.
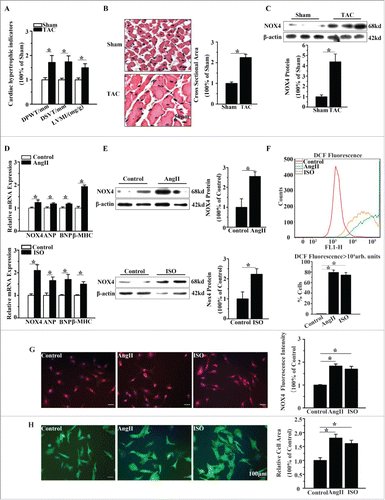
Furthermore, we determined NOX4 expression in primary cultured NCMs treated with hypertrophic agonists (AngII 10−5M or ISO 10−5M for 24h). Congruent with the in vivo results, the protein expression of NOX4 in NCMs treated with AngII or ISO were elevated 2.5-fold and 2.2-fold compared with untreated controls, respectively (). And the hypertrophic markers atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MHC) were moderately increased in cardiomyocytes treated with AngII or ISO (). Flow cytometry (FCM) analysis showed that, the number of dichlorofluorescein(DCF) positive cells in AngII or ISO group was markedly increased compared with that in the control group, indicating that ROS generation was significantly increased in AngII or ISO groups, in parallel with the increase in NOX4 expression (). In addition, immunofluorescence staining demonstrated that NOX4 expression in NCMs treated with AngII or ISO was approximately increased 2-fold compared with that in controls (). The surface areas of NCMs exposed to AngII or ISO was also increased about 2-fold compared with that of controls ().
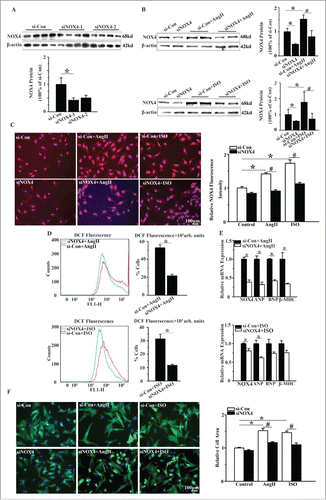
We then evaluated whether NOX4 promoted cardiomyocyte hypertrophy induced by AngII or ISO. As shown in , genetic silencing of NOX4 inhibited the elevation of NOX4 expression in NCMs exposed to AngII or ISO by up to 70% and reduced the number of DCF positive cells by about 50%. Moreover, NOX4 silencing significantly decreased the surface areas and the expression of the hypertrophic markers in NCMs exposed to AngII or ISO (). These results indicated that NOX4 and NOX4-derived ROS play a critical role in the hypertrophic stimuli-induced cardiomyocyte hypertrophy.
Figure 2. Silencing of NOX4 by specific siRNA ameliorated cardiomyocyte hypertrophy in vitro. (A) Western blotting analysis for NOX4 in NCMs transfected with siRNA. si-Con, siNOX4-1 and siNOX4-2 group indicate negative control group, NOX4 siRNA groups respectively. * indicates P<0.05 vs. si-Con. (B) Western blotting analysis for NOX4 protein expression in NCMs in the presence or absence of AngII(10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom) after transfected with si-Con or siNOX4. * indicates P<0.05 vs. si-Con; # indicates P<0.05 vs. si-Con+AngII or si-Con+ISO. (C) Immunofluorescence staining for NOX4 expression in NCMs in the presence or absence of AngII(10−5M for 24h) or ISO (10−5M for 24h) after transfected with si-Con or siNOX4. * indicates P<0.05 vs. si-Con; # indicates P<0.05 vs. si-Con+AngII or si-Con+ISO. (D) FCM analysis for ROS generation in NCMs in the presence or absence of AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom) after transfected with si-Con or siNOX4. * indicates P<0.05 vs. si-Con+AngII or si-Con+ISO. (E) mRNA expression of NOX4 and hypertrophic markers in NCMs in the presence or absence of AngII (10−5M for 24h, Top) or ISO(10−5M for 24h, Bottom) after transfected with siNOX4. * indicates P<0.05 vs. si-Con+AngII or si-Con+ISO. (F) Surface area determination of NCMs treated with AngII (10−5M for 24h) or ISO (10−5M for 24h) after transfected with si-Con or siNOX4. * indicates P<0.05 vs. si-Con; # indicates P<0.05 vs. si-Con+AngII or si-Con+ISO. n = 4 for A-F. The fluorescent micrograph is representative of cells from 4 independent visual fields.
NOX4 activates Xbp1s in hypertrophic stimuli-induced myocardial hypertrophy
In order to investigate whether Xbp1s is a potential signaling molecule through which NOX4 promotes the cardiomyocyte hypertrophy, we first studied the effects of NOX4 on ERS signaling in NCMs. As shown in , protein levels of Grp78, Xbp1s and ATF4, as ERS markers, were significantly increased in the heart. However, protein level of ATF6 was unaltered. Consistent with our observation in vivo, the levels of Xbp1s expression were elevated 4 to 5 folds in AngII or ISO-treated NCMs compared with that in untreated controls. Expression of ATF4 was elevated nearly 2 folds in NCMs treated with AngII or ISO (). Gene silencing of NOX4 in NCMs treated with AngII or ISO inhibited the upregulation of Grp78 and Xbp1s by up to 60% and 80%, respectively (). These results indicated that Xbp1s was activated by NOX4 upon hypertrophic stimuli.
Figure 3. NOX4 activated Xbp1s in cardiac hypertrophy. (A) Western blotting analysis for Xbp1s in rats 4 weeks after TAC or sham surgery. n = 6. * indicates P<0.05 vs. sham-operated group. (B) Western blotting analysis for Xbp1s in NCMs treated with AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom). n = 4. * indicates P<0.05 vs. control. (C) Western blotting analysis for Xbp1s in NCMs in the presence or absence of AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom) after transfected with si-Con or siNOX4. n = 4. * indicates P<0.05 vs. si-Con; # indicates P<0.05 vs. siNOX4+AngII or siNOX4+ISO. (D) Western blotting analysis for Xbp1s signaling in NCMs of indicated groups. NCMs treated with AngII (10−5M for 24h), ISO (10−5M for 24h) or H2O2 (100μM for 2h) showed significantly higher activation of Xbp1 compared to blank group. The activation of Xbp1 was restored by pretreatment with either NAC (10mM for 1h) or GKT137831(5μM for 1h). n = 3. *, ## indicates P<0.05 vs. blank group and DMSO group, respectively; ** indicates P<0.05(AngII, ISO, H2O2 group vs. AngII+NAC, ISO+NAC, H2O2+NAC group, respectively); # indicates P<0.05(AngII and ISO group vs. AngII+GKT137831 and ISO+ GKT137831group, respectively). (E) Western blotting analysis for Xbp1 and NOX4 in NCMs transfected with si-Con or siXbp1(1–3). n = 3. * indicates P<0.05 vs. si-Con group. (F) Surface area determination of NCMs in the presence or absence of AngII (10−5M for 24h) or ISO (10−5M for 24h) after transfected with si-Con or siXBP1. n = 4. The fluorescent micrograph is representative of cells from 4 independent visual fields.
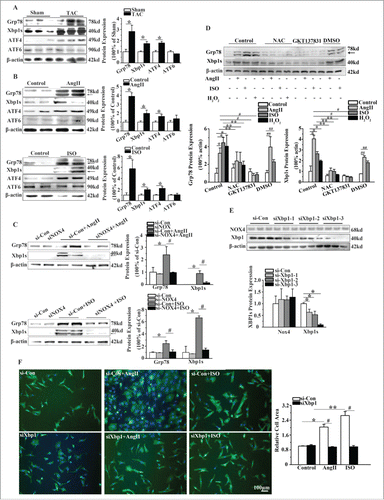
Because the primary ROS product of NOX4 is H2O2, we used H2O2, N-acetyl-L-cysteine, (NAC, a ROS scavenger) and GKT137831 (a NOX4 inhibitor) to assess whether NOX4 regulated above-described signaling pathways through its ROS product. As demonstrated in , the protein level of Grp78 and Xbp1s in NCMs exposed to H2O2 was elevated 4-fold and 2-fold, respectively, compared with that in NCMs in control group. Moreover, NAC and GKT137831 significantly blunted the upregulation of Grp78 and Xbp1s in NCMs treated with AngII or ISO().
We next determined whether activation of Xbp1s signaling was responsible for cardiomyocyte hypertrophy. As expected, the surface areas of NCMs transfected with Xbp1 siRNA in the presence of AngII or ISO were decreased by almost 50% compared with the cells transfected with negative control siRNA (). These observations suggest that activation of Xbp1s signaling is essential to hypertrophic stimuli-induced cardiomyocyte hypertrophy.
NOX4 activates the RIPK1-related NF-κB signaling pathways in hypertrophic stimuli-induced myocardial hypertrophy
In the heart tissue of TAC rats, the expression of RIPK1 was increased by almost 3.5 folds compared with that of sham-operated rats (). Moreover, NF-κB was activated, as shown by the increased phosphorylation of P65 subunit of NF-κB (). In NCMs, AngII or ISO upregulated the expression of RIPK1 and activated NF-κB signaling by strikingly 5 to 7 folds (). Notably, gene silencing of NOX4 by specific siRNA effectively inhibited the upregulation of RIPK1 and phosphorylation of P65 subunit of NF-κB by up to 70% in NCMs treated with AngII or ISO (), indicating that NOX4 is required for the activation of downstream RIPK1 and NF-κB signaling pathway.
Figure 4. NOX4 activated RIPK1-related NF-κB signaling pathways in cardiac hypertrophy. (A) Western blotting analysis for RIPK1 and phosphorylated P65 subunit of NF-κB in rats 4 weeks after TAC or sham surgery. n = 6. * indicates P<0.05 vs. sham-operated group. (B) Western blotting analysis for RIPK1 and phosphorylated P65 subunit of NF-κB in NCMs treated with AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom). n = 4. * indicates P<0.05 vs. control. (C) Western blotting analysis for RIPK1 and phosphorylated P65 subunit of NF-κB in NCMs in the presence or absence of AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom) after transfected with si-Con or siNOX4. n = 4. * indicates P<0.05 vs. si-Con, # indicates P<0.05 vs. si-Con+AngII or si-Con+ISO. (D) Western blotting analysis for RIPK1 related NF-κB signaling in NCMs. NCMs treated with AngII (10−5M for 24h), ISO (10−5M for 24h) or H2O2 (100μM for 2h) showed significantly higher activation of RIPK1/NF-κB signaling compared to blank group. The activation of RIPK1/NF-κB signaling was restored by pretreatment with either NAC (10mM for 1h) or GKT137831(5μM for 1h). n = 3. *, ## indicates P<0.05 vs. blank and DMSO group, respectively; ** indicates P<0.05(AngII, ISO and H2O2 group vs. AngII+NAC, ISO+NAC and H2O2+NAC group, respectively); # indicates P<0.05(AngII and ISO vs. AngII+GKT137831 and ISO+ GKT137831 group, respectively). (E) Western blotting analysis for RIPK1 and phosphorylated P65 subunit of NF-κB in NCMs in the presence or absence of AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom) after transfected with si-Con or siRIPK1. n = 4. *, ** indicates P<0.05 vs. si-Con, # indicates P<0.05 vs. si-Con+AngII or si-Con+ISO. (F) Immunoprecipitation analysis for K63-linked polyubiquitination of RIPK1. RIPK1 immunoprecipitates from whole-cell lysates of NCMs were subjected to Western blotting. NCMs treated with AngII (10−5M for 24h) or ISO (10−5M for 24h) showed significantly higher K63-linked polyubiquitination of RIPK1 as compared to control group. The polyubiquitination of RIPK1 was restored by transfection of si-Con or siNOX4 into NCMs treated with AngII (10−5M for 24h) or ISO (10−5M for 24h). (G) Western blotting analysis for RIPK1 and phosphorylated P65 subunit of NF-κB in NCMs treated with AngII (10−5M for24h, Top) or ISO (10−5M for 24h, Bottom) after pretreatment of Nec-1(10nM). n = 4. * indicates P<0.05 vs. control. (H) Surface area determination of NCMs in the presence or absence of AngII(10−5M for 24h) or ISO (10−5M for 24h) after transfected with si-Con or siRIPK1. n = 4. The fluorescent micrograph is representative of cells from 4 independent visual fields. * indicates P<0.05 vs. si-Con; # indicates P<0.05 vs. si-Con+AngII or si-Con+ISO.
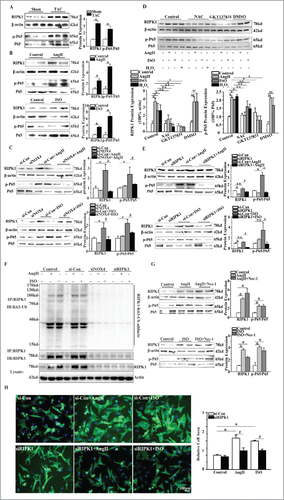
We also tested whether the RIPK1-related NF-κB signaling was activated in NCMs exposed to H2O2, and deactivated by NAC and GKT137831. As demonstrated in , P65 subunit phosphorylation of NF-κB was enhanced in NCMs exposed to AngII, ISO or H2O2, in line with the elevated protein level of RIPK1. Furthermore, NAC blunted the P65 subunit phosphorylation of NF-κB and upregulation of RIPK1 in NCMs treated with AngII, ISO or H2O2. Similar results were obtained in NCMs pretreated with GKT137831().
As RIPK1 functions dichotomously either as a scaffold protein to activate NF-κB signaling or as a kinase to induce necroptosis, we then employed siRIPK1 and RIPK1 kinase inhibitor necstatin-1(Nec-1) to clarify the specific role of elevated RIPK1 in the development of cardiomyocyte hypertrophy induced by AngII or ISO. Interestingly, we observed that under AngII or ISO stimulation, siRIPK1 significantly inhibited phosphorylation of P65 subunit of NF-κB and in turn mitigated cardiomyocyte hypertrophy ( and ). We further determined the K63(lysine 63)-linked polyubiquitination of RIPK1. The polyubiquitination level of RIPK1 was enhanced in NCMs treated with AngII or ISO, in keeping with the enhanced protein level of RIPK1 (). As well, after genetic silencing of NOX4 or RIPK1, the RIPK1 polyubiquitination reduced as the protein level of RIPK1 was markedly decreased (). By contrast, Nec-1 pretreatment (10nM) did not inhibit the expression of RIPK1 and the phosphorylation of P65 subunit of NF-κB in NCMs stimulated with AngII or ISO (). These findings provided evidence that RIPK1 acts as a “scaffold protein” in a kinase-independent mode.
Activation of the RIPK1-related NF-κB pathway is mediated by Xbp1s
Finally, we tested whether Xbp1s regulates the RIPK1-related NF-κB signaling pathway. As indicated in , in the presence of AngII or ISO, gene silencing of Xbp1s significantly reduced the upregulation of RIPK1 by almost 60% and its downstream phosphorylation of P65 subunit of NF-κB by approximately 70%.
Figure 5. RIPK1-related NF-κB signaling pathway was regulated by Xbp1s. (A) Western blotting analysis for RIPK1 and phosphorylation of P65 subunit of NF-κB in NCMs in the presence or absence of AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom) after transfected with of si-Con or siXbp1. n = 3. *, ** indicates P<0.05 vs. si-Con, # indicates P<0.05 vs. si-Con+AngII or si-Con+ISO. (B) Western blotting analysis for ATF4 and ATF6 in NCMs treated with AngII (10−5M for 24 h, Top) or ISO (10−5M for 24h, Bottom) after transfected with siNOX4. n = 4. * indicates P<0.05 vs. si-Con+AngII. (C) Western blotting analysis for ATF4 in NCMs transfected with si-Con or siATF4(1–3). n = 3. * indicates P<0.05 vs. si-Con group. (D) Western blotting analysis for RIPK1 and phosphorylation of P65 subunit of NF-κB of in NCMs in the presence or absence of AngII (10−5M for 24h, Top) or ISO (10−5M for 24h, Bottom) after transfected with of si-Con or siATF4. n = 3. * indicates P<0.05 vs. si-Con group.
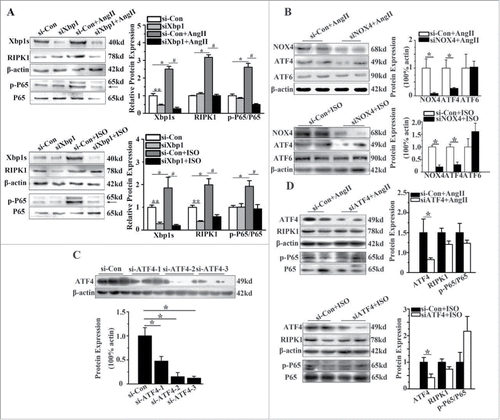
As mentioned previously, the protein level of ATF4 was also increased in NCMs treated with AngII or ISO. Therefore, we tested whether ATF4 plays a role as similar as Xbp1s. The protein level of ATF4 exhibited significant decline after genetic silencing of NOX4. However, after ATF4 knockdown with ATF4 siRNA, the RIPK1 and P65 subunit phosphorylation of NF-κB demonstrated no significant change in NCMs treated with AngII or ISO (-), indicating that the RIPK1-related NF-κB pathway is just activated by Xbp1s.
Discussion
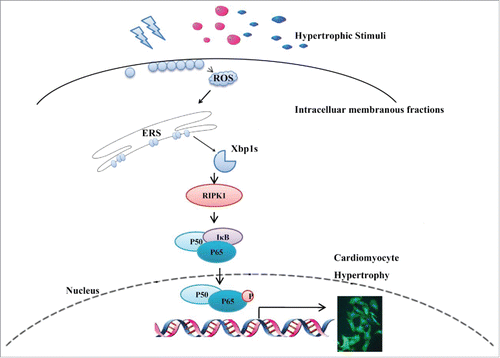
In the present study, we have demonstrated a novel signaling pathway underlying the NOX4-induced cardiac hypertrophy. We found that: (1) Splicing of Xbp1 accounts for NOX4-induced cardiac hypertrophy, and (2) Xbp1s activated downstream RIPK1-related NF-κB signaling to cause cardiac hypertrophy ().
Figure 6. Schematic representation of the Xbp1s/RIPK1/NF-κB pathway by which NOX4 triggered cardiac hypertrophy. Upon hypertrophic stimuli(pressure overload caused by TAC, neurohumoral factors AngII and ISO), elevated NOX4 expression and ROS generation resulted in splicing of Xbp1, enhanced RIPK1 expression and ensuing phosphorylation of P65, ultimately led to cardiomyocyte hypertrophy.
Consistent with previous reports,Citation25-27 our experiments have demonstrated that NOX4 is upregulated by various vasoactive agonists such as AngII and ISO, and in response to pressure overload. However, some data from mice with NOX4 transgene have shown that overexpression of NOX4 directly cause cell apoptosis without significant hypertrophy,Citation28,29 The possible reason for this disparity between our data and theirs might be explained by the different amount of NOX4 expression. In our study, the expression of NOX4 is increased 4.2-, 2.5-, and 2.2-fold by TAC, AngII and ISO, respectively. However, in contrast to the relatively modest elevation of NOX4 expression under pathological stimulation, cardiac-specific overexpression of NOX4 causes a 10-fold increase in NOX4 abundance and 8-fold increase in ROS levels. Based on this disparity, we speculate that excessive NOX4 and its derivative ROS rapidly and directly cause cardiomyocyte death, while the relatively moderate elevation of NOX4 expression and its derivative ROS lead to cardiomyocyte hypertrophy.
One of the major findings of the present study is that enhanced Xbp1 splicing contributes to the development of NOX4-related cardiomyocyte hypertrophy. As a powerful transcription factor, spliced Xbp1 exerts strong prosurvival effects under various conditions, including cardiac hypoxic conditions, Citation30 cardiac ischemia/reperfusion injury,Citation31 and cell proliferation.Citation32-35 All these reports indicate that Xbp1s may act as a pro-proliferative signaling molecule to mediate the cardiomyocyte hypertrophy in response to stresses. Interestingly, a recent study further found that Xbp1s contributes to the progression of cardiac hypertrophy through regulating cardiac angiogenesis,Citation10 in support of our finding that Xbp1 is critical for cardiac hypertrophy. Besides, our study revealed that cardiac expression of Xbp1s is regulated by NOX4 and NOX4-related ROS. Nonetheless, the precise mechanism by which NOX4 induces splicing of Xbp1 is not explored in our work. Based on the observation that expression of ERS markers like Grp78 and ATF4 were elevated in vivo and in vitro, we postulated that NOX4 and NOX4-related ROS in ER rather than in other intracellular compartments caused ERS and ensuing Xbp1 spicing. However, further studies are necessary to elucidate the detailed mechanism by which NOX4 activates Xbp1s in cardiomyocyte hypertrophy.
Another major finding of the present study is that Xbp1s causes cardiac hypertrophy through activation of the RIPK1-related NF-κB signaling. Although RIPK1 mediates cell survival and death, many studies in recent years focus on its role in induction of programmed necrosis (necroptosis) by various cellular stresses.Citation11,12 Likewise, recent cardiovascular researches concerning RIPK1 primarily focus on its role in induction of necroptosis in ischemia/reperfusion(I/R) injury and heart failure,Citation13-15 and only a few studies point out that RIPK1 is involved in the TGF-β-induced activation of cardiac fibroblasts.Citation36 In our previous study, we found that RIPK1 mediates the palmitic acid (PA)-induced cardiomyocyte hypertrophy, and inhibition of RIPK1 by Nec-1 attenuates cardiomyocyte hypertrophy induced by PA.Citation18 Indeed, in the present study, we found that RIPK1 is important for hypertrophic growth of cardiomyocytes undergoing ERS induced by hypertrophic stimuli (pressure overload/AngII/ISO). Nonetheless, we further found that RIPK1 acts as a scaffold protein to activate NF-κB signaling in a kinase-independent mode. This was supported by the observations that elevated RIPK1 protein level and its polyubiquitination in NCMs treated with AngII or ISO were blunted by the genetic inhibition of NOX4 or ISO while cannot be inhibited by Nec-1. By contrast with PA-induced cardiomyocyte hypertrophy, cardiomyocyte hypertrophy induced by hypertrophic stimuli (pressure overload/AngII/ISO) does not require the kinase activity of RIPK1. These seemingly contradictory results might be attributed to the differential stresses which might represent 2 distinct clinical entities. In other words, typical hypertrophic stimuli (pressure overload/AngII/ISO) represent hypertrophy induced by mechanical stress and related activation of neurohumoral systems, while PA represents lipotoxicity-related hypertrophy. In brief, our study revealed that the RIPK1 related NF-κB signaling acts as a novel alternative pathway of UPR signaling to induce cardiac hypertrophy. This finding bears practical implication. On one hand, RIPK1 is involved in cardiomyocyte loss of I/R injury and heart failure via mediating necroptosis or apoptosis.Citation13 On the other hand, it participates in the hypertrophic growth of cardiomyocytes through NF-κB pathways. Based on these facts, when RIPK1 is considered as a therapeutic target, differential strategy should be adopted to target its diverse roles in different clinical settings.
In conclusion, our study provides evidence that upregulation of NOX4 promotes cardiomyocyte hypertrophy induced by hypertrophic stimuli through Xbp1s/RIPK1/NF-κB signaling pathway. These data offer insights into novel mechanisms with potential therapeutic implications for myocardial hypertrophy.
Materials and methods
Antibodies and reagents
Antibodies were obtained from the following commercial sources: anti-NOX4 (1:1000, NB 110–58849) was purchased from Novus; Anti-Grp78(1:500, #3183), anti-ATF4(1:500, D4B8), anti-p65(1:1000, C22B4), anti-phosphorylated p65(1:1000, 3033S) and anti-RIPK1(1:1000, D94C12) were purchased from Cell Signaling Technology; Anti-Xbp1s(1:500, ab37152), anti-ATF6(1:500, ab11909) and anti-linkage-specific K63 ubiquitin(1:1000, ab179434) was purchased from Abcam; anti-β-actin was purchased from Santa Cruz; anti-α-actinin(1:200, A7811) were purchased from Sigma Aldrich; Angiotensin II(A9525), isoproterenol (I5627), and N-acetyl-L-cysteine(A7250) were purchased from Sigma Aldrich; Necrostatin-1(S8037) and GKT137831(S717101) were obtained from Selleckchem.
Animal model of transverse aortic constriction (TAC)
Sprague-Dawley(SD) rats with pressure overload-induced cardiac hypertrophy were created by TAC for 4 weeks.Citation6 Rats were anesthetized with pentobarbital sodium (50 mg/kg i.p.) and entered into the abdominal cavity. The abdominal aorta above the bifurcation of left renal artery and aorta was banded by a 5–0 silk suture ligature tied firmly against an 18-gauge needle. The needle was removed to yield a constriction of 1.2 mm in diameter. Sham-operated rat underwent a similar surgical procedure without banding of the aorta. 4 weeks after surgery, the rats were lightly anesthetized with pentobarbital sodium (30–50 mg/kg i.p.) and subjected to echocardiography. Then the hearts were dissected and snap-frozen in liquid nitrogen and stored at −80°C. All animals used in this study received humane care and the study was approved by the ethics committee of Sichuan University (ethic number 2014003A).
Echocardiography analysis
Two-dimensional (2D) guided M-mode echocardiography was performed. Parasternal long-axis views, short-axis views, and 2D guided M-mode images of short axis at the mid-papillary muscle level were recorded. The LVESD (LV end-systolic diameter) and LVEDD (LV end-diastolic diameter), diastolic and systolic LV wall thickness, diastolic posterior wall thickness (DPWT) and diastolic ventricular septal thickness (DVST) were measured in 3 consecutive cardiac cycles. LVMI (Left ventricular mass index) was calculated as the ratio of whole heart weight to body weight.
Histological analysis
Hearts were excised and washed with saline solution. Then the heart tissues were fixed with 10% formalin, embedded in paraffin and cut transversely close to the apex. After rehydration, the sections (4–5 μm) of heart were obtained and mounted onto slides and stained with haematoxylin-eosin (HE). After staining, images of cardiomyocytes were captured by microscopy and the cross sectional areas of the cardiomyocytes were determined by a quantitative digital analysis system (Image J, National Institutes of Health). More than 100 myocytes in the sections from at least 6 different rat samples were calculated in each group.
Isolation and primary culture of neonatal cardiomyocytes (NCMs)
NCMs were harvested from the myocardium of Sprague-Dawley neonatal rats 0–3d after birth.Citation6 Rat ventricles were quickly removed and cut into tiny pieces, then digested with a mixture of 0.05% trypsin solution (Gibco) and 0.05% collagenase II(Gibco). Isolated cells from each digestion were pooled in DMEM (Hyclone Laboratories) supplemented with 10% fetal bovine serum (biological industries), penicillin (100 U/ml), streptomycin (100 U/ml) and Bromodeoxyuridine (0.1 mM). Then the cell suspensions were centrifuged at 1000rpm for 10 min, the isolated cells were re-suspended and seeded in a culture flask. After 90 min for fibroblast adherence, cardiomyocytes were isolated and seeded onto 6-well or 12-well plates. 24 hr later, isolated NCMs were serum-starved for 24h before treatment with hypertrophic stimuli: ISO (10−5M) or AngII(10−5M) for 24h.
Small interfering RNA (siRNA) transfection
Cells were seeded on 6-well or 12-well plates at 50–70% confluence before transfection. Individual siRNAs(100nM, Invitrogen),Citation37 lipofectamine 3000 (Invitrogen) and DMEM were mixed and incubated at room temperature for 15 min. siRNA-lipofectamine 3000 complexes were added to cells and 24 hr later AngII (10−5M) or ISO (10−5M) was added into cells. All siRNA sequences are shown in .
Table 1. Oligonucleotide sequences for siRNAs.
Intracellular ROS measurement
Intracellular ROS production was determined by oxidative conversion of cell permeable 2′,7′-dichlorofluorescein diacetate (H2DCFDA; Sigma-Aldrich) to fluorescent dichlorofluorescein. NCMs were washed with serum-free DMEM and incubated with H2DCFDA (10μM) for 60 min at 37°C in incubator. Cells were then trypsinized (0.05% trypsin free of EDTA, Gibco), resuspended in PBS and the samples were analyzed by flow cytometry (FCM, BD FACSCalibur) at an excitation and emission wavelengths of 485 nm and 530 nm, respectively.
Immunofluorescent staining and determination of cell surface area
NCMs seeded on 12-well plates were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. After blocking with 1% BSA cells were incubated with mouse anti-α-actinin (1:200) for surface area determination or rabbit anti-NOX4 (1:200) overnight at 4°C. After a brief wash, cells were incubated with an anti-mouse Alexa Fluo 488-conjugated secondary antibody (1:500, invitrogen) or anti-rabbit Alexa Fluo 594-conjugated secondary antibody (1:500, invitrogen). After the antibody solution was removed, the cells were counterstained for nuclei with DAPI (1:1000, Sigma Aldrich) for another 10 min. Finally, cells were mounted and visualized with an inverted microscope (Nikon). NCMs from 4 independent microscopic fields were outlined for surface area determination and NOX4 fluorescence intensity analysis in each group.
Protein extraction, immunoblotting and immunoprecipitation
For tissue extraction, the frozen heart tissue was homogenized and lysed by a RIPA buffer (Byotime) with protease and phosphatase inhibitor cocktail, subsequently sonicated for 15s. For whole cell extraction, cells were lysed with RIPA buffer (Byotime) with protease and phosphatase inhibitor cocktail. After centrifugation at 16000×g for 10min (4°C), the protein concentrations were subsequently determined using the BCA Protein Assay Kit (ThermoFisher scientific). Then the protein samples (50μg) were subjected to SDS-PAGE, transferred to a PVDF membrane (Millipore, Bedford, MA), blocked with 5% skim milk and probed with corresponding primary antibodies and HRP-conjugated secondary antibodies, followed by detection with Enhanced Chemiluminescence (ECL, Bio-Rad). The protein levels were normalized to β-actin. For immunoprecipitation, whole cell lysates were mixed and precipitated with RIPK1(1:100) and protein A/G Agarose beads by incubation at 4°C. The bound proteins were removed by boiling in SDS buffer and resolved in SDS-PAGE gels for immunoblotting analysis.
Real-time polymerase chain reaction analysis (qRT-PCR)
The total RNA was harvested from frozen left ventricle tissues or cell lysates using TRIzol (Invitrogen). 1μg RNA of each sample was used to reverse-transcribe into cDNA using the ReverTra Ace qPCR RT Master Mix kit (Toyobo). PCR was performed using a SYBR® Green master mixes (Bio-Rad). All primer details were shown in . The mRNA levels were normalized to β-actin.
Table 2. Primer sequences for qRT-PCR.
Statistical analysis
All data are presented as mean ± SEM. Data were analyzed with SPSS17.0 software. Differences between groups were evaluated for significance using single-tailed Student's t-test of unpaired data or one-way analysis of variance (ANOVA). Bonferroni post-test analysis was used to compensate for multiple testing procedures. A value of P < 0.05 was considered significant.
Disclosures of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Natural Science Foundation of China under Grant No.11072163 and Grant No.11372204; and the Science and Technology Department of Sichuan Province under Grant No. 2013FZ0005.
References
- Schiattarella GG, Hill JA. Inhibition of hypertrophy is a good therapeutic strategy in ventricular pressure overload. Circulation 2015; 131:1435-47; PMID:25901069; http://dx.doi.org/10.1161/CIRCULATIONAHA.115.013894
- Park CS, Cha H, Kwon EJ, Sreenivasaiah PK, Kim DH. The chemical chaperone 4-phenylbutyric acid attenuates pressure-overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem Biophys Res Commun 2012; 421:578-84; PMID:22525677; http://dx.doi.org/10.1016/j.bbrc.2012.04.048.
- Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal 2009; 11:2409-27; PMID:19388824; http://dx.doi.org/10.1089/ars.2009.2625.
- Sag CM, Santos CX, Shah AM. Redox regulation of cardiac hypertrophy. J Mol Cell Cardiol 2014; 73:103-11; PMID:24530760; http://dx.doi.org/10.1016/j.yjmcc.2014.02.002.
- Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Ann Rev Physiol 2013; 75:49-67; PMID:23020580; http://dx.doi.org/10.1146/annurev-physiol-030212-183707.
- Lu WW, Zhao L, Zhang JS, Hou YL, Yu YR, Jia MZ, Tang CS, Qi YF. Intermedin1-53 protects against cardiac hypertrophy by inhibiting endoplasmic reticulum stress via activating AMP-activated protein kinase. J Hypertens 2015; 33:1676-87; PMID:26136070; http://dx.doi.org/10.1097/hjh.0000000000000597.
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012; 13:89-102; PMID:22251901; http://dx.doi.org/10.1038/nrm3270.
- Wu R, Zhang QH, Lu YJ, Ren K, Yi GH. Involvement of the IRE1alpha-XBP1 pathway and XBP1s-dependent transcriptional reprogramming in metabolic diseases. DNA Cell Biol 2015; 34:6-18; PMID:25216212; http://dx.doi.org/10.1089/dna.2014.2552.
- Jiang D, Niwa M, Koong AC. Targeting the IRE1alpha-XBP1 branch of the unfolded protein response in human diseases. Semin Cancer Biol 2015; 33:48-56; PMID:25986851; http://dx.doi.org/10.1016/j.semcancer.2015.04.010.
- Duan Q NL, Wang P, Chen C, Yang L, Ma B, Gong W, Cai Z, Zou MH, Wang DW. Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell 2016; 15:625-33; http://dx.doi.org/doi:10.1111/acel.12460
- Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol 2015; 25:347-53; PMID:25662614; http://dx.doi.org/10.1016/j.tcb.2015.01.001
- Murphy JM, Silke J. Ars Moriendi; the art of dying well - new insights into the molecular pathways of necroptotic cell death. EMBO Rep 2014; 15:155-64; PMID:24469330; http://dx.doi.org/10.1002/embr.201337970.
- Li L, Chen Y, Doan J, Murray J, Molkentin JD, Liu Q. Transforming growth factor beta-activated kinase 1 signaling pathway critically regulates myocardial survival and remodeling. Circulation 2014; 130:2162-72; PMID:25278099; http://dx.doi.org/10.1161/CIRCULATIONAHA.114.011195.
- Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res 2014; 103:206-16; PMID:24920296; http://dx.doi.org/10.1093/cvr/cvu146.
- Zhang T, Zhang Y, Cui M. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med 2016; 22:175-82; PMID:26726877; http://dx.doi.org/10.1038/nm.4017.
- Luan Q, Jin L, Jiang CC, Tay KH, Lai F, Liu XY, Liu YL, Guo ST, Li CY, Yan XG, et al. RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy. Autophagy 2015; 11:975-94; PMID:26018731; http://dx.doi.org/10.1080/15548627.2015.1049800.
- Estornes Y, Aguileta MA, Dubuisson C, De Keyser J, Goossens V, Kersse K, Samali A, Vandenabeele P, Bertrand MJ. RIPK1 promotes death receptor-independent caspase-8-mediated apoptosis under unresolved ER stress conditions. Cell Death Dis 2014; 5:e1555; PMID:25476903; http://dx.doi.org/10.1038/cddis.2014.523.
- Zhao M, Lu L, Lei S, Chai H, Wu S, Tang X, Bao Q, Chen L, Wu W, Liu X. Inhibition of receptor interacting protein kinases attenuates cardiomyocyte hypertrophy induced by palmitic acid. Oxidat Med Cell Longev 2016; 2016:1451676; PMID:27057269; http://dx.doi.org/10.1155/2016/1451676.
- Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A 2010; 107:15565-70; PMID:20713697; http://dx.doi.org/10.1073/pnas.1002178107
- Maejima Y, Kuroda J, Matsushima S, Ago T, Sadoshima J. Regulation of myocardial growth and death by NADPH oxidase. J Mol Cell Cardiol 2011; 50:408-16; PMID:21215757; http://dx.doi.org/10.1016/j.yjmcc.2010.12.018.
- Eletto D, Chevet E, Argon Y, Appenzeller-Herzog C. Redox controls UPR to control redox. J Cell Sci 2014; 127:3649-58; PMID:25107370; http://dx.doi.org/10.1242/jcs.153643.
- Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol 2010; 30:3553-68; PMID:20457808; http://dx.doi.org/10.1128/MCB.01445-09.
- Santos CX, Hafstad AD, Beretta M, Zhang M, Molenaar C, Kopec J, Fotinou D, Murray TV, Cobb AM, Martin D, et al. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF2alpha-mediated stress signaling. EMBO J 2016; 35:319-34; PMID:26742780; http://dx.doi.org/10.15252/embj.201592394.
- Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, Marie JC, Pouzet C, Samadi M, Elbim C, et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol 2004; 24:10703-17; PMID:15572675; http://dx.doi.org/10.1128/mcb.24.24.10703-10717.2004.
- Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie LH, Tian B, Sadoshima J. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ Res 2013; 112:651-63; PMID:23271793; http://dx.doi.org/10.1161/circresaha.112.279760.
- Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res 2003; 93:802-5; PMID:14551238; http://dx.doi.org/10.1161/01.res.0000099504.30207.f5.
- Zeng SY, Chen X, Chen SR, Li Q, Wang YH, Zou J, Cao WW, Luo JN, Gao H, Liu PQ. Upregulation of Nox4 promotes angiotensin II-induced epidermal growth factor receptor activation and subsequent cardiac hypertrophy by increasing ADAM17 expression. Can J Cardiol 2013; 29:1310-9; PMID:23850346; http://dx.doi.org/10.1016/j.cjca.2013.04.026.
- Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation 2015; 131:643-55; PMID:25589557; http://dx.doi.org/10.1161/circulationaha.114.011079.
- Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 2010; 106:1253-64; PMID:20185797; http://dx.doi.org/10.1161/circresaha.109.213116.
- Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res 2006; 99:275-82; PMID:16794188; http://dx.doi.org/10.1161/01.RES.0000233317.70421.03
- Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014; 156:1179-92; PMID:24630721; http://dx.doi.org/10.1016/j.cell.2014.01.014.
- Bhandari P, Song M, Dorn GW, 2nd. Dissociation of mitochondrial from sarcoplasmic reticular stress in Drosophila cardiomyopathy induced by molecularly distinct mitochondrial fusion defects. J Mol Cell Cardiol 2015; 80:71-80; PMID:25555803; http://dx.doi.org/10.1016/j.yjmcc.2014.12.018.
- Iwakoshi NN, Lee AH, Glimcher LH. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev 2003; 194:29-38; PMID:12846805
- Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008; 134:743-56; PMID:18775308; http://dx.doi.org/10.1016/j.cell.2008.07.021.
- Hess DA, Humphrey SE, Ishibashi J, Damsz B, Lee AH, Glimcher LH, Konieczny SF. Extensive pancreas regeneration following acinar-specific disruption of Xbp1 in mice. Gastroenterology 2011; 141:1463-72; PMID:21704586; http://dx.doi.org/10.1053/j.gastro.2011.06.045.
- Song J, Zhu Y, Li J, Liu J, Gao Y, Ha T, Que L, Liu L, Zhu G, Chen Q, et al. Pellino1-mediated TGF-beta1 synthesis contributes to mechanical stress induced cardiac fibroblast activation. J Mol Cell Cardiol 2015; 79:145-56; PMID:25446187; http://dx.doi.org/10.1016/j.yjmcc.2014.11.006.
- Jiang Q, Fu X, Tian L, Chen Y, Yang K, Chen X, Zhang J, Lu W, Wang J. NOX4 mediates BMP4-induced upregulation of TRPC1 and 6 protein expressions in distal pulmonary arterial smooth muscle cells. PloS One 2014; 9:e107135; PMID:25203114; http://dx.doi.org/10.1371/journal.pone.0107135.