
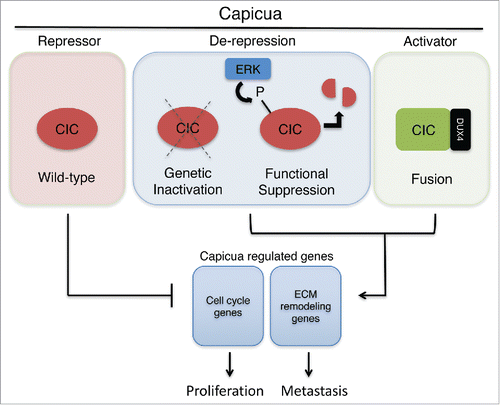
Capicua (CIC) is an evolutionary conserved transcriptional repressor that regulates terminal differentiation through MEK-ERK mediated gene expression in the head and tail of Drosophila.Citation1 Thus, Capicua gains its name from Catalan translation, “head” and “tail.” Through development of an in vivo orthotopic lung cancer model, we recently revealed that genetic inactivation of CIC through deletion or loss-of-function mutation can de-repress pro-metastatic effectors, ETV4 and MMP24, which are necessary and sufficient for metastasis.Citation2 Moreover, we found that activation of MEK-ERK signaling leads to post-translational suppression of CIC through rapid protein degradation (CitationFig. 1). Collectively, our data suggest that hyperactivation of MAPK signaling, a hallmark in many human cancers, may enhance metastatic potential via ERK-driven suppression of CIC that promotes ETV4-MMP24 upregulation.
MEK-ERK activation, which is present in ∼60% of lung adenocarcinomas results in rapid CIC protein degradation, which may in part explain the high rate of metastatic recurrence and poor survival in patients with early stage lung adenocarcinoma that undergo curative resection.Citation3,4 Thus, CIC loss and increased ETV4 or MMP24 expression in earlier stage lung adenocarcinoma patients are putative biomarkers of metastatic recurrence, and can potentially identify a subset of patients who may benefit from more aggressive primary, neo-adjuvant, or adjuvant therapy. Collectively, our findings suggest that RAF-MEK-ERK inhibitor treatment can restore CIC protein expression to block primary or secondary metastatic progression in patients by dampening ETV4-MMP24 levels in cancers with genetically-intact CIC.
The anti-metastatic effects of MAPK signaling inhibition may be distinct from, or in addition to, an impact on cancer growth, as CIC may regulate cancer dissemination and potentially tumor growth via distinct transcriptional repertoires. Specifically, we found that the CIC effector, MMP24, regulated metastatic capacity, but did not significantly impact cancer cell growth. Yet, when we performed a genetic rescue of CIC, specifically in CIC deficient cells, tumor growth and cell cycle regulatory genes were suppressed. To add an additional layer of complexity, we did not observe a significant increase in cancer cell growth despite an increase in metastatic efficiency when endogenous CIC was genetically silenced. These findings argue that CIC transcriptional control of different cellular phenotypes is tightly regulated and potentially dose-dependent. This specialization in CIC functional output may allow for specificity and context-dependence during normal embryogenesis and malignant cancer progression.
Further functional dissection of CIC regulated pathways may reveal additional therapeutic targets. In gastric adenocarcinoma and oligodendroglioma, for example, where the predominant mode of CIC inactivation is through genetic alteration (mutation or copy number loss), MAPK inhibition is unlikely to provide a therapeutic benefit. Thus, targeting alternative CIC regulated pathways, including cell cycle or the CIC target gene, ETV4, may potentially limit tumor growth and progression in these cancers. A third mode of regulation that results in aberrant CIC expression is in the context of an oncogenic fusion. In contrast to the repressor activity of wild-type CIC, the CIC-DUX4 fusion results in activation of conserved CIC target genes, including ETV1, ETV4, and ETV5.Citation5,6 Intriguingly, the CIC-DUX4 fusion results in high ETV4 expression in undifferentiated small round cell sarcoma, a highly aggressive, chemotherapy-insensitive tumor that typically presents with synchronous metastasis and poor survival.Citation7 These clinical findings suggest that the CIC-DUX4 fusion may activate the same conserved downstream pathways that we uncovered in lung and gastric adenocarcinoma tumors with functional or genetic loss of CIC. Whether the transcriptional landscape, and thus therapeutic targets in CIC-DUX4 sarcoma are identical to tumors with genetic inactivation of CIC remains unknown and will be the focus of future studies.
In summary, our in vivo dissection of lung cancer metastasis uncovered a CIC-regulated developmental pathway that is co-opted in human cancer to promote tumor progression and metastasis. While the mechanism of CIC deregulation in histologically different cancers may indeed be tissue-specific, the convergence upon a core set of highly conserved pathways acting downstream of CIC provides tremendous opportunity for therapeutic exploitation to limit cancer metastasis.
Figure 1. Wild-type Capicua (CIC) represses pro-metastatic and cell cycle genes. Genetic inactivation or post-translational suppression of CIC can de-repress a core set of CIC-regulated genes to promote highly conserved functional phenotypes. In the context of the CIC-DUX4 fusion, CIC is transformed into an activator of target genes.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Jiménez G1, Guichet A, Ephrussi A, Casanova J. Relief of gene repression by torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev 2000; 14:224-31; PMID:10652276
- Okimoto RA, Breitenbuecher F, Olivas VR, Wu W, Gini B, Hofree M, Asthana S, Hrustanovic G, Flanagan J, Tulpule A, et al. Inactivation of Capicua drives cancer metastasis. Nat Genet 2017; 49(1):87-96; PMID:27869830; http://dx.doi.org/10.1038/ng.3728
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511:543-50; PMID:25079552; http://dx.doi.org/10.1038/nature13385
- Winton T, Livingston R, Johnson D, Rigas J, Johnston M, Butts C, Cormier Y, Goss G, Inculet R, Vallieres E, et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N Engl J Med 2005; 352:2589-97; PMID:15972865; http://dx.doi.org/10.1056/NEJMoa043623
- Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, Gotoh T, Motoi T, Fukayama M, Aburatani H, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 2006; 15:2125-37; PMID:16717057; http://dx.doi.org/10.1093/hmg/ddl136
- Specht K, Sung YS, Zhang L, Richter GH, Fletcher CD, Antonescu CR. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 2014; 53:622-33; PMID:24723486; http://dx.doi.org/10.1002/gcc.22172
- Le Guellec S, Velasco V, Pérot G, Watson S, Tirode F, Coindre JM. ETV4 is a useful marker for the diagnosis of CIC-rearranged undifferentiated round-cell sarcomas: a study of 127 cases including mimicking lesions. Mod Pathol 2016; 29:1523-31; PMID:27562494; http://dx.doi.org/10.1038/modpathol.2016.155