
ABSTRACT
The IgA receptor, Fcar (CD89) consists of 5 sequence segments: 2 segments (S1, S2) forming the potential signal peptide, 2 extracellular EC domains that include the IgA binding site, and the transmembrane and cytoplasmic tail (TM/C) region. Numerous Fcar splice variants have been reported with various combinations of the sequence segments mentioned above. Here, we report a novel splice variant termed variant APD isolated from a healthy volunteer that lacks only the IgA-binding EC1 domain. Despite possessing the complete signal peptide S1+S2, the variant APD is only found in the intracellular space whereas the wild-type variant 1 is efficiently secreted and variant 4 leaks to the extracellular space. Further mutational experiments involving signal peptide replacements, cleavage site modifications, and studies on alternative isoforms demonstrate that despite the completeness of the signal peptide motif, the presence of the EC1 domain is essential for efficient extracellular export.
Introduction
Human Immunoglobulin A (Ig) Fc receptor (CD89) or Fcar binds to both human IgA1 and 2 Fc regions and plays an important role in mucosal immunity.Citation1,2 It was first cloned from U937 monocyte cell line in 1990Citation3 and 8 protein-coding splice variants have since been reported ()Citation4-8 in addition to possible non-coding RNA transcripts. From the protein sequence point of view, Fcar consist of 5 distinct sequence segments formed by 5 exons, namely S1, S2, EC1, EC2, and TM/C. The crystal structures of the soluble extracellular (EC) domains of Fcar are available.Citation9-12 The S1 and S2 exons translate to the potential cleavable signal peptide,Citation5 the EC1 and EC2 exons deliver the extracellular domains (EC1 binds IgA Fc), and the TM/C exon is responsible for the transmembrane and cytoplasmic tail region.Citation3
Table 1. A summary table of existing Fcar variants including our novel variant APD. Accessions, references and exon segment features are listed for comparison. Corresponding amino acids of the sequence segments are numbered based on the full-length variant 1. Each variant had the signal peptide analyzed using SignalP with scores: max C, mean S, D score and possible cleavage site calculations shown. The extracellular detection of the variants are expressed in percentage derived from FACS data as shown in .
Distinct from other immunoglobulin Fc receptors (FcRs), Fcar binds IgA Fc at a 2:1 ratio, causing the IgA Fc binding EC1Citation12 (binding kinetics of 0.96 × 106 M−1 contributed by R82 and H85Citation13) to bend almost perpendicularly to EC2. In addition, Fcar is co-localized at chromosome 19 with the leukocyte immunoglobulin-like receptors (LIR/LILR, ILTs) and killer inhibitory receptors (KIRs) but not with the other Ig FcRs (FcγRs or FcϵR)Citation9,12 that are found on chromosome 1. Nonetheless, functionally, Fcar bears greater similarity to the FcRs.
Of the Fcar RNA transcripts reported in the literature,Citation4,14,15 most lack known function and detectable protein isoforms. For example, variant 9 (a splice variant that lacks the TM/C domain,Citation16 accession number NM_133279.2 recently removed from the database) is speculated to negatively regulate secreted IgA binding isoforms.Citation4 In the case of variant 7 (a splice variant that lacks the S2 and EC1 domain,Citation4 see ), the protein is not found on the outer membrane nor does it bind IgA. Nonetheless, the functions of these splice variants remain unknown.
In the process of producing purified Fcar protein from the cDNA from a healthy human donor, we discovered a new, previously unreported Fcar splice variant (named APD) that has all the Fcar sequence segments, with the exception of the EC1 domain. Surprisingly, we found that this variant when expressed by recombinant means, is kept intracellularly without export. Elaborate mutational studies of the signal peptide region and beyond as well as studies of other Fcar variants showed that even a complete S1+S2 signal peptide together with cleavage site is insufficient for efficient export and the presence of the EC1 domain is instrumental for this purpose.
Results
PCR based detection of Fcar variants
Using Fcar specific primers, we performed PCR amplification repeatedly on fresh cDNA from peripheral blood mononuclear cells (PBMC) isolated from a volunteer. We chose the smallest band of ∼700 bp for TOPO cloning and sequencing. This analysis led to the discovery of a new, previously unreported Fcar variant that had all the known domains, with the exception of EC1 (accession: KT805280, deposited at GenBank).
In , we provide comprehensive information about the 8 protein-coding splice variants of Fcar together with that about our new variant APD. Sequence alignment of these isoforms () showed that they differed predominantly in 2 locations: (1) in the S1 + S2 region; and (2) in the EC1 domain (). For example, variant 4 lacks only the S2 segment, while variant 7 lacks both S2 and EC1. Comparing the amino acid sequences of the APD variant with variants 1, 4 and 7 (), the new variant lacks only the EC1 domain.
Figure 1. Fcar variants and variant APD. (A) Multiple sequence alignment of the current 8 variants with variant APD. The drawing above the alignment shows the secondary structure presentation of the wild type Fcar_v1. The blocks represent helices; arrows for β strands; and lines for coils. (B) A schematic presentation of the 9 variants showing the domains: signal peptide domains (S1 in blue and S2 in red), extracellular domains (EC1 in green, EC2 in purple), transmembrane (TM/C in orange), and the secreted region (in black) present only in Fcar_v6. Among the 4 variants 1, 4, 7 and APD, both 4 and 7 lack the S2 domain, whereas variants 7 and APD lack the EC1 domain. (C) Agarose gel image of the PCR product from the cDNA template obtained from a healthy volunteer's PBMC using Fcar_vAPD specific primers that bind to S2 and EC2 domains. The band sizes of 580bp (600 bp in the gel) and 289bp (307 bp in the gel) were calculated using GelApp.Citation30

To rule out sequencing and cloning artifacts, we designed primers specific to the S2 and EC2 domains to re-screen the PBMC cDNA library. Due to the placement of the primers, only the full-length Fcar variant 1, and variant APD should be detected. As shown in , this is indeed the case and only bands corresponding to the size of variant 1 and APD were observed.
Sequence analysis of Fcar protein variants including APD using ANNIECitation17 (this software environment includes SignalPCitation18 together with other protein sequence analysis tools) reaffirmed that the S1 and S2 segments form the predicted signal peptide only when both are present (). This suggests that S1 alone may be insufficient for extracellular membrane localization.
Confocal microscopy studies on localization of Fcar variants
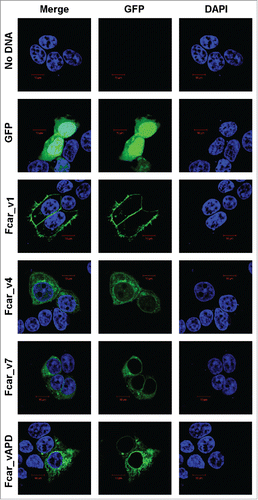
We cloned variants 1, 4, 7 and APD with C-terminal GFP after the TM/C domain to avoid interfering with the N-terminal signal peptide function and performed confocal microscopy to study the variants in transfected cells (). Analysis of the transfected HEK293 EXPI cells and the controls showed that the eGFP-transfected cells (negative control) to be detected throughout the intracellular space. Similarly, variants 4, 7, and APD were also found to be located in intracellular space. Only variant 1 is observed to have GFP predominantly lining at the plasma membrane of the cells. The result is surprising since APD contains a fully predicted signal peptide; yet, it is not exported to the extracellular membrane.
Figure 2. Confocal microscopy analysis of the Fcar cellular localization with GFP-tagged proteins and DAPI. EXPI cells were transfected with plasmids containing Fcar variants 1, 4, 7, and APD with c-terminal eGFP. Controls: plasmid (e-GFP only) and no plasmid. Cells were fixed and mounted onto glass slides and viewed using Zeiss confocal microscopy Meta upright LSM5100. Positive control eGFP proteins as well as Fcar variants 4, 7, and APD were found to be intracellular localized. Only Fcar_v1 (Fcar_v1-GFP) was found to be predominantly lining the plasma membrane. The figure shown is representative of at least 4 independent experiments.
Flow cytometric analysis of extracellular Fcar
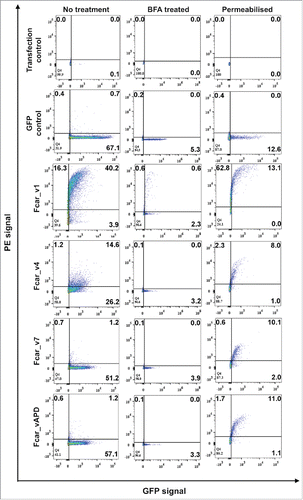
As an alternative method to study secretion, we performed FACS analyses on the variant-GFP transfected cells (, ). For additional detection of variants lacking EC1 (the typical epitopes for anti-CD89 antibodies are located in EC1), mouse polyclonal anti-CD89 followed by anti-mouseIg antibody-PE were used. Of 3 different primary commercial polyclonal and monoclonal antibodies tested (data not shown), only one could detect variants without EC1. The eGFP control cells, while present, did not show anti-CD89-PE staining, neither did the cells transfected with variants other than variant 1. The variant 1-GFP (positive control) showed significant (∼40%) PE and GFP staining. Interestingly, the variant 4-GFP transfected cells showed some “leakiness” with small amounts (∼14%) of extracellular anti-CD89-PE and GFP staining whereas variants 7 and APD had <2% dual positive cells (, left column). To ensure that the lack of PE staining was not due to the lack of EC1 epitopes, cell-permeabilized polyclonal mice anti-CD89 followed by anti-mouse-Ig-PE staining was performed and showed similar levels of detection of Fcar intracellularly and in agreement to the GFP fusion protein detection (, right column). With agreement from both the GFP and anti-CD89 antibodies, the outcome of the experiments is unexpected as variant 4 without a full signal peptide sequence (absence of S2) is found to leak to the extracellular space (as detected by PE) when variant APD (having the full signal peptide sequence) does not get exported in measurable quantities.
Figure 3. FACS analysis of the Fcar variants with GFP tags. EXPI 293 cells transfected with the various Fcar-eGFP variants were stained with mouse polyclonal anti-CD89 antibody followed by anti-mouse IgG1-PE. Values on the dot plots represent percentages of cells in each quadrant. The left column shows the experiment set for “no treatment” to detect protein expression on the cell surface. The center column shows BFA treated cells. The right column shows permeabilised cell staining to detect intracellularly proteins. Gating was performed based on the no plasmid and eGFP controls. eGFP control cells were transfected with e-GFP construct and stained with polyclonal anti-CD89 followed by anti-mouse IgG conjugate with PE. All variants with the exception of the wild-type variant 1-GFP (40.2%) were found to be in minute amounts on the cell surface as detected by the PE signals. Dampening of all Fcar variants-GFP expression were observed with BFA treatment. When untreated, Fcar_v4 transfected cells showed 14.6% extracellular presence. This effect was also diminished with BFA treatment. All variants with the GFP tags were detected by polyclonal anti-CD89 when cells were permeabilised. Each FACS experiment is based on 20,000 events and of at least 4 independent experiments.
Confirmation of secretory pathway involvement
To investigate the mechanism of Fcar exportation to the membrane, we incubated the transfected cells with Brefeldin A (BFA) to inhibit secretion via the Golgi Apparatus (GA). FACS analysis showed a significant decrease in detection of all Fcar variants including variant 1 (, center column). Thus, Fcar is indeed exported via the Endoplasmic Reticulum (ER)-GA secretory pathway.
Analysis of mutations in the signal peptide motif
To understand the reasons for the paradoxical behavior of variants 4 and APD, we carried a series of mutational experiments in the signal peptide motif region of Fcar variants (). First, we hypothesized that the possible insufficiency of the cleavage site region in APD might prevent export despite the presence of a fully predicted signal peptide. To note, the max C score of SignalP for APD is lower than those for other Fcar variants (). In a first series of these experiments, we performed post-signal peptide cleavage site mutations where the first 2 amino acids of EC2 (Leucine – Tyrosine or LY) were substituted by site-directed mutagenesis (SDM) to Aspartic acid and Phenylalanine (DF) to resemble the N-terminal of EC1. Indeed, the max C score of this mutant is improved (). Nevertheless, we found no significant change in the extracellular staining for the mutated variants APD and 7 (lacking both S2 and EC1) as observed in the FACS analysis ().
Table 2. Comparison of Fcar variants and the signal peptide and cleavage site mutants used in this study. Table shows the protein residue number, and various mutations made to the respective exon segments. The variants and mutants were analyzed using the SignalP tool for calculations on the signal peptide (max C, mean S, D score) and possible cleavage sites. Extracellular detection percentages were determined from FACS experiments as show in where variant 1 and variant APD were repeatedly tested.
Figure 4. Analysis of signal peptide and cleavage site mutations. EXPI 293 cells transfected with the various mutants modifications made to the Fcar-eGFP variants and stained with mouse polyclonal anti-CD89 antibody followed by anti-mouse IgG1-PE for FACS analysis. Values on the dot plots represent percentages of cells in each quadrant. Gating was performed based on the no plasmid and eGFP controls. eGFP control cells were transfected with e-GFP construct and stained with polyclonal anti-CD89 followed by anti-mouse IgG conjugate with PE. (A) Analysis of variant 7 and APD mutants with the first 2 amino acids in EC2 (LY) substituted with that of EC1 (DF). With and without the S2 domain, modifications to the start of EC2 did not result in increased cell surface presence of the variants. (B) Analysis of signal peptide and cleavage site mutations. Fcar mutants with the IgE signal peptide with and without the additional S2-EC1 cleavage site QE were used to displace the original S1+S2 signal peptide on variants 1 and APD as well as mutant variant APD with EC1 (DF) substitution mutations on EC2. Regardless of the signal peptide and the presence of the QE cleavage site and the substitution on EC2, variant APD was not detected to have increased extracellular presence as determined by FACS. Each FACS experiment is based on 20,000 events and repeated at least 3 independent times.
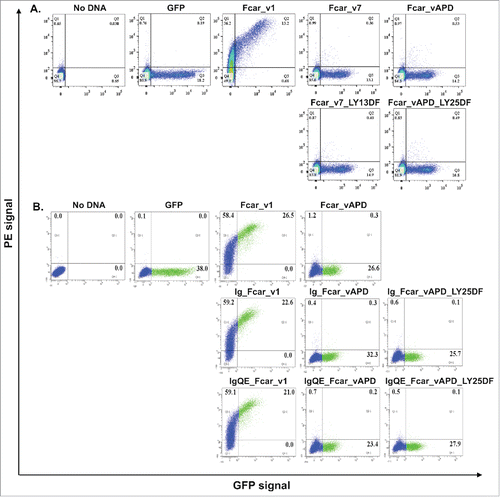
Second, we hypothesized that the S1+S2 might represent a weak signal peptide sequence. Analysis of variant 4 using SignalP showed that the S1 domain alone with EC1 to have a low D score value of 0.181 (). Together with S2, the score increased to 0.626 for variant 1 (with EC1 following S2) and to 0.599 for variant APD (with EC2 following S2). When we replaced the S1+S2 sequence with the signal peptide of human IgE (i.e. MDWTWILFLVAAATRVHS), the similar analysis resulted in a higher score of > 0.7 (0.737 when this sequence was followed by EC1, and 0.706 when by EC2). To validate this computational result, we cloned the human IgE signal peptide in the place of S1 and S2 in the cases of variant 1 and variant APD. In addition, we performed the same clones yet with additional 2 amino acids “QE” (which are the last 2 residues of S2) right after the IgE signal peptide sequence because we suspected that these 2 residues might contribute to the cleavage region of the S1+S2 signal peptide as in variant 1. To facilitate cleavage, we further mutated the first 2 amino acids of EC2 (i.e., LY) of variant APD to DF (mimicking EC1). These products were named accordingly in as Ig_Fcar_v1, Ig_Fcar_vAPD, Ig_Fcar_vAPD_LY25DF, IgQE_Fcar_v1, IgQE_Fcar_vAPD, and IgQE_Fcar_vAPD_LY25DF (see also ). FACS analysis of these signal peptide mutants showed surprising results (). We found that only the Ig_Fcar_v1 and IgQE_Fcar_v1 mutant GFP fusion proteins get efficiently exported when all the other mutants remained intracellularly localized (, ).
The results imply that any efficient signal peptide motif in front of Fcar protein variants alone is insufficient to force export of Fcar. Once the EC1 domain is included, secretion to the extracellular space is facilitated. The presence of the EC1 domain is even sufficient to compensate for a bad signal peptide as the absence of S2 in variant 4 nevertheless allowed leakiness to the extracellular side of the plasma membrane.
Discussion
In this paper, we report the discovery and characterization of a new variant of Fcar that specifically lacked only the EC1 domain compared with the other known isoforms. We have termed this newly discovered variant as “APD.” We initially hypothesized variant APD to function as an IgA-independent regulator (see review on Fcar activityCitation14) because it contains the complete S1-S2 signal peptide domains (see ), EC2 and TM/C domains (to activate ITAM). However, confocal microscopy and FACS analyses showed this variant APD to be absent from extracellular membrane localization (). With an intact TM and EC2, it could have been able to bind to FcR γ-chain and activate the immunoreceptor tyrosine-based activation motif (ITAM) signaling pathwayCitation14 in an IgA-independent manner. Similarly, the great variation of S2 and EC1 occurrences among Fcar variants in combination with other sequence regions commands enquiries about their functional role. Nonetheless, excessive expression of this variant APD over variant 1 is likely to result in poor response to IgA and deficient mucosal adaptive immunity.
The surprising finding in this work is the observation of Fcar APD being unable to be secreted via the signal peptide secretory pathway despite apparently complete signal peptides (both S1+S2 and human IgE leader) together with a cleavage site region. To note, the functional signal peptides were also recognized by sequence analytic tools that find the signal peptide motif (). Originally, we hypothesized that the signal peptide might be the reason that caused the failure of extracellular membrane dispatch of variant APD. The S1+S2 domain was computationally detected as a signal peptide only when both S1 and S2 were present. This result agrees with a previous studyCitation4 where S1+S2 domain formed a helical structure and only functioned as a signal peptide when both were present. Since variant 4 lacked the S2 domain and had compromised membrane localization when compared with variant 1, the hypothesis was strongly supported.
This observation is even more surprising in the context of “leakiness” of Fcar variant 4 that has just an incomplete leader peptide (without S2) with remnant signal peptide qualities only. Notably, the S1 sequence has a low signal peptide D score of 0.181 (see ). It is unlikely that antibodies entering cellsCitation19 would account for the observed leakage of variant 4 in contrast to variants 7 and APD. To note, all the 3 variants obtained relatively similar levels of anti-CD89-PE staining in permeabilization experiments. The export of variant 4 is dependent on Golgi apparatus as the “leakiness” disappeared in the presence of BFA (). We acknowledge that our permeabilization and BFA experiments reflected lower cell counts despite efforts to increase the cell counts at transfection. This low cell count was due to the harsh permeabilization treatment and the protein secretion inhibition by BFA that had led to increased cell bursting and cell death, respectively. Nonetheless, we have shown that the variants were expressed in similar amounts as determined by both GFP and mouse anti- human CD89 followed by anti-mouse Ig-PE.
We performed many mutational experiments to improve the efficiency of the signal peptide at the N-terminus of variant APD. Firstly, we enhanced the cleavage site region (the Max C score and D score are lower compared with other variants, ) without achieving successful export. Notably, higher scores of signal peptide were obtained when followed by EC1 over EC2 (0.626 vs 0.599); therefore, the first 2 amino acids of EC2 (i.e., LY) were displaced with the first 2 amino acids of EC1 (i.e., DF). However, variant APD remained largely undetected on the extracellular membrane.
Finally, we completely replaced the signal peptide with that of human IgE leader, both with and without the enhanced cleavage site regions with substitutions and/or insertions (). Yet, this still failed to achieve any notable extracellular localization. Since computational analysis suggested that the natural Fcar S1+S2 signal peptide to be a weak signal peptide, we further replaced the signal peptide with that of IgE. Regardless of the additional insertion of the Fcar S2-EC1 cleavage site amino acids (QE) after the IgE signal peptide cleavage site (VHS) with and without the first 2 amino acids of EC2 substituted, we were still unable to detect significant variant APD on the extracellular membrane. Considering the findings of our signal peptide and cleavage site experiments, the whole EC1 is shown to be important for extracellular localization in a manner that can partially compensate for the weaker S1 alone signal peptide (shown in the leakiness of variant 4).
Thus, our results indicate that, for extracellular localization of Fcar variants, the presence of the EC1 domain seems more important than the completeness of the motif and the efficiency of the signal peptide. Though rarely observed, there are cases described in the literature where apparently functional signal peptides are insufficient to force secretion of proteins.Citation20 Current evidence supports the ideas about differentially efficient signal peptides, the role of subsequent sequences and domains, as well as their mutual interactions.
The phenomenon of EC1-dependent secretion of Fcar deserves further attention. While EC1 is crucial for the correct extracellular localization; without S2, the export efficiency was significantly decreased (as seen for variant 4). Structural simulation studies made this observation plausible. One possible explanation is that EC1 bridges and prevents non-specific interactions between the hydrophobic stretch of the signal peptide and hydrophobic patchesCitation21,22 on the EC2 domain. Results of explicit solvent molecular dynamics simulation using AMBER14 and normal mode analysis using Bio3d package,Citation23,24 for the variants 1 and APD demonstrated different fluctuations of the signal peptides with respect to EC2 and/or EC1. In the cases of variant 1, the S1+S2 domain oscillated around EC1 and EC2 (∼40–50 Å distant from EC2) whereas it was rigid and maintained close proximity (∼15–20 Å) to the EC2 in variant APD. We observed hydrophobic interactions of the Leucine-rich regions between S1+S2 domain and EC2 may explain the retention of the signal peptide in the hydrophobic groove of EC2 formed by residues Tyr105 and Leu69. This interaction is mitigated by the presence of EC1 which bridged and prevented the non-specific interactions between these hydrophobic patches (see Fig. S1B).
To conclude, the relative levels of protein expression of Fcar isoforms and the functions of their non-exported variants remain unknown. The lack of extracellular localization makes it unlikely for them to play an IgA-independent regulatory role to interact with the membrane bound proteins for signal transduction. Nevertheless, the study of variant APD and those of variants 1, 4 and 7 have revealed that Fcar is exported in a GA-dependent manner and that the IgA-binding EC1 domain is essential for correct extracellular localization of Fcar.
Materials and methods
Fcar variant 1 gene synthesis
Fcar variant 1 gene (accession no: NM002000.3) was synthesized by Blue Heron Biotech LLC (USA). Human IgE signal peptide (Genbank accession J00227) was cloned from previously obtained plasmids.Citation25
Fcar variant APD isolation from PBMCs
The novel Fcar variant APD (accession no: KT805280) was cloned from a volunteer with informed consent (ethics approval from Singhealth cIRB ref: 2013/540/D). Human PBMC were extracted from blood using Ficoll® Paque Plus according to manufacturer's recommendations (Cat no: 17–1440–02, GE Healthcare). Total RNA was extracted from PBMC using Trizol Reagent (Cat no: 15596026, Life Technologies) and cDNAs were synthesized using Tetro cDNA synthesis kit (Cat no: Bio-65042, Bioline), all used according to the manufacturer's recommendations. PCR products were cloned using Q5 High Fidelity DNA polymerase (Cat no: M0491L, NEB) and Fcar-Cloning primers (Forward: 5′-ATG GAC CCC AAA CAG ACC ACC CTC-3′; Reverse: 5′-CTT GCA GAC ACT TGG TGT TCG TGC A-3′), followed by analysis on 1% agarose gels (Quintech Life Sciences Pte Ltd). Bands of the desired sizes were determined using GelAppCitation26 and cloned using TOPO PCR cloning kit (Cat no: 450245, Life Technologies), according to manufacturer's recommendations.
C-terminally eGFP-linked Fcar variants
Fcar variants 1 and APD were cloned into pEGFP-N3 (Clonetech) using XhoI and BamHI sites incorporated to the PCR products with overhanging primers: XhoI_Fcar F (5′-GTC ATC TCG AGA TGG ACC CCA AAC AGA CC-3′) and Fcar_BamHI R (5′- GCT AGG ATC CCT TGC AGA CAC TTG GTG-3′). The plasmids were transformed into chemically competent DH5α Fcar variants 4 and 7 (ascension numbers: NM_133272.3 and NM_133277.3) were obtained from site directed mutagenesis (SDM) of variants 1 and APD by removing exon 2 (S2 domain). SDM primer pairs used are: 1) FcarV4mut F (5′-GAC CAC CCT CCT GTG TCT TGG GGA CTT TCC CAT G-3′) and FcarV4mut R (5′- CAT GGG AAA GTC CCC AAG ACA CAG GAG GGT GGT C-3′); and 2) FcarV7mut F (5′-CAC CCT CCT GTG TCT TGG CTT GTA TGG CAA ACC C-3′) and FcarV7mut R (5′-GGG TTT GCC ATA CAA GCC AAG ACA CAG GAG GGT GGT C-3′).
Fcar variants with EC2 to EC1 (LY to DF) substitution mutations
Mutant Fcar variants APD and 7 with LY (EC2) to DF (EC1) mutations were created using SDM. SDM primers used for variant APD are FcarVAPD_LY25DF F (5′-GGC ACA GGA AGG CGA TTT CGG CAA ACC CTT C-3′) and FcarVAPD_LY25DF R (5′-GAA GGG TTT GCC GAA ATC GCC TTC CTG TGC C-3′). For variant 7, the primers used are: FcarV7_LY13DF F(5′-CCT GTG TCT TGG CGA TTT TGG CAA ACC CTT C-3′) and FcarV7_LY13DF R (5′-GAA GGG TTT GCC AAA ATC GCC AAG ACA CAG G-3′).
Fcar variants with IgE signal peptide with and without additional QE cleavage sites
Fcar variants with the IgE signal peptide were formed by joining the IgE signal peptide from previously obtain plasmidsCitation25 with the Fcar variants with and without EC1 and ligated into pEGFP. Primers for PCR of IgE signal peptide were IgE_SP_F (5′-GAA GCT AGC ATG GAC TGG ACC TGG ATC-3′) and IgE_SP_R (5′-GGA GTG CAC TCG AGT GGC TGC-3′). Primers for PCR of Fcar variants were FcarV1_IgE_F (5′-CAA CTC GAG TGC ACT CCG GGG ACT TTC CCA TG-3′), FcarVAPD_IgE_F (5′- CCA CTC GAG TGC ACT CCG GCT TGT ATG GCA AAC −3′); FcarVAPD_DF_IgE_F (5′- CCA CTC GAG TGC ACT CCG GCG ATT TCG GCA AAC-3′). Primers for PCR of Fcar variants with additional cleavage site (QE) were FcarV1_QE_IgE_F (5′-CCA CTC GAG TGC ACT CCC AGG AAG GGG ACT TTC-3′); FcarVAPD_QE_IgE_F (5′-CAA CTC GAG TGC ACT CCC AGG AAG GCT TGT ATG GCA A-3′); FcarVAPD_DF_QE_IgE_F (5′-CCA CTC GAG TGC ACT CCC AGG AAG GCG ATT TCG GCA A-3′). One reverse primer was used for PCR of all 6 Fcar variant mutants - Fcar_R (5′-GTT GGT ACC CTT GCA GAC ACT TGG TG-3′).
All plasmids for transfection were extracted using Quintech plasmid miniprep kitCitation27 (Cat no: QT100–1101, Quintech Lifesciences Pte Ltd). All cloning steps were sequence validated using DNAappCitation28 and analyzed using MAFFT,Citation29 ANNIE,Citation17 and SignalP 4.0.Citation18
Confirmation of Fcar variant APD in volunteer PBMC
Confirmation detection of Fcar variant APD in PBMC was performed using FcarVAPDID primers (F: 5′-GCC AGA GGA TTC AGG CAC AG-3′ and R: 5′-TTA CTG GGG AAG GAC CAC AG-3′). The PCR product was analyzed as performed above.
Florescent activated cell sorting (FACS) analysis
HEK293 EXPI293 (Life Technologies) cells were seeded at 3 × 105 cells/well in 6-well plates or 1.8 × 106 cells/dish in 10 cm culture plates. Transfection of cells were performed (described previouslyCitation30) on the following day with extra media with or without Brefeldin A (2.5ug/ml) added after 3 hours. Cells were harvested after 15 hours and stained with antibodies. Cells were first washed with cold PBS and scraped in the presence of FACS buffer (PBS with 10% FBS, 1% Sodium azide). They were spun down and the pellet was incubated with mouse polyclonal anti-Fcar/CD89 (Cat no: LS-C307768, Life Span Biosciences) for 30 minutes at 4°C followed by anti mouse-IgG1-PE (Cat no: 130–098–106, Mitenyi) for 30 minutes at 4°C. These were followed by 3 washes of FACS buffer in between the antibodies binding steps. For the cell permeabilization experiments, addition incubation step of 0.1% TritonX in PBS for 5 minutes at 4°C during harvest of cells was performed before staining. FACS analysis was done using FACSAria IIu SORP (Becton-Dickinson) at Biopolis Shared Facilities, A*STAR, Singapore.
Confocal microscopy
HEK293 EXPI293 cells were seeded at 7 × 104 cells/well on glass coverslips on 12-well plates and transfected the following day as described above. Cells were washed with PBS, fixed with 2% formaldehyde and mounted onto glass slides with Vectashield, Hard SET Mounting Medium with DAPI (Cat no: H-1500, Vector Laboratories). Images were taken at 64x with upright confocal system, LSM 510 META (Carl Zeiss) at Biopolis Shared Facilities, A*STAR, Singapore.
Computational analysis of the signal peptides
SignalP 4.0 Citation18 (Organism group: Eukaryotes, default D-cutoff values) was used to predict the occurrence of N-terminal signal leader peptides. Sequences with calculated D scores in the range of [0.450–1.000] are predicted to carry a signal peptide.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
WHL drafted the manuscript and performed the confocal microscopy and FACS staining experiments; WLL cloned the new variant Fcar APD; JYY performed the signal peptide cloning and experiments; BE and FE performed the sequence analyses, contributed to the assessment of the results and the writing of the manuscript; CSV, CTTS and SKEG discussed computational prediction and mutational analyses; and CTTS assisted in the figures. SKEG supervised the whole project. All authors read and approved the manuscript.
KCCY_A_1281480_supplementary_material.tif
Download TIFF Image (3 MB)Acknowledgments
We would like to thank Mr Chan Xiang Wei for his assistance in the laboratory preparation.
Funding
This work was supported by the JCO1334i00050 grant from Joint Council Office, Agency for Science, Technology, and Research (A*STAR) in Singapore.
Related Research Data
References
- Herr A, White C, Milburn C, Wu C, Bjorkman P. Bivalent binding of IgA1 to FcαRI suggests mechanism for cytokine activation of IgA phagocytosis. J Mol Biol 2003; 327:645-57; PMID:12634059; http://dx.doi.org/10.1016/S0022-2836(03)00149-9
- Maliszewski C, Vanden Bos T, Shen L, Schoenborn M, Kubagawa H, Beckmann M, Monteiro R. Recombinant soluble IgA Fc receptor: generation, biochemical characterization, and functional analysis of the recombinant protein. J Leukoc Biol 1993; 53:223-32; PMID:8454945
- Maliszewski C, March C, Schoenborn M, Gimpel S, Shen L. Expression cloning of a human Fc receptor for IgA. J Exp Med 1990; 172:1665-72; PMID:2258698; http://dx.doi.org/10.1084/jem.172.6.1665
- Van Dijk T, Bracke M, Caldenhoven E, Raaijmakers J, Cammers J, Koenderman L, de Groot R. Cloning and characterization of Fc alpha Rb, a novel Fc alpha receptor (CD89) isoform expressed in eosinophils and neutrophils. Blood 1996; 88:4229-38; PMID:8943858
- Reterink T, Verweij C, van Es L, Daha M. Alternative splicing of IgA receptor (CD78) transcripts. Gene 1996; 175:279-80; PMID:8917112; http://dx.doi.org/10.1016/0378-1119(96)00152-7
- Pleass R, Andrews P, Kerr M, Woof J. Alternative splicing of the human IgA receptor CD89 in neutrophils and eosinophils. Biochem J 1996; 318:771-7; PMID:8836118; http://dx.doi.org/10.1042/bj3180771
- Patry C, Sibille Y, Lehuen A, Monteiro R. Indentification of Fcα Receptor (CD89) isoforms generated by alternative splicing that are differentially expressed between blood monocytes and alveolar macrophages. J Immunol 1996; 156:4442-8; PMID:8666819
- Morton H, Schiel A, Janssen S, van de Winkel J. Alternatively spliced forms of the human myeloid Fcα receptor (CD89) in neutrophils. Immunogenetics 1996; 43:246-7; PMID:8575829; http://dx.doi.org/10.1007/s002510050057
- Yang M, Yao M, Gao G, Wang L, Zhang W, Rao Z. Crystal structure of the ectodomain of human FcαRI. J Biol Chem 2003; 278:27966-70; PMID:12783876; http://dx.doi.org/10.1074/jbc.C300223200
- Yang M, Xu G, Li S, Sun L, Shi N, Zeng W, Pang H, Zhang W, Rao Z. Crystallization and preliminary crystallographic analysis of the extracellular fragment of FcαRI/CD89. Acta Crystallog Section D 2003; 59:2251-3; ; http://dx.doi.org/10.1107/S0907444903020602
- Wenig K, Sendermann P. Purification, crystallization and X-ray diffraction analysis of the extracellular part of the human Fc receptor for IgA, FcαRI (CD89). Acta Crystallog Section D 2003; 59:2247-50; ; http://dx.doi.org/10.1107/S0907444903016421
- Herr A, Ballister E, Bjorkman P. Insights into IgA-mediated immune responses from the crystal structures of human FcαRI and its complex with IgA1-Fc. Nature 2003; 423:614-20; PMID:12768205; http://dx.doi.org/10.1038/nature01685
- Wines B, Hulett M, Jawieson G, Trist H, Spratt J, Hogarth P. Identification of residues in the first domain of the human Fcα receptor essential for interaction with IgA. J Immunol 1999; 162:2146-53; PMID:9973489
- Monteiro R, van de Winkel J. IgA Fc receptors. Annu Rev Immunol 2003; 21:177-204; PMID:12524384; http://dx.doi.org/10.1146/annurev.immunol.21.120601.141011
- Van Vuuren A, van Egmond M, Coenen M, Morton H, van de Winkel J. Characterization of the human myeloid IgA Fc receptor I (CD89) gene in a cosmid clone. Immunogenetics 1999; 49:586-9; PMID:10380711; http://dx.doi.org/10.1007/s002510050544
- Toyabe S, Kuwano Y, Takeda K, Uchiyama M, Abo T. IgA nephropathy-specific expression of the IgA Fc receptors (CD89) on blood phagocytic cells. Clin Exp Immunol 1997; 110:226-32; PMID:9367406; http://dx.doi.org/10.1111/j.1365-2249.1997.tb08321.x
- Ooi H, Kwo C, Wildpaner M, Sirota F, Eisenhaber B, S M-S, Wong W, Scheleiffer A, Eisenhaber F, Schneider G. ANNIE: integrated de novo protein sequence annotation. Nucleic Acids Res 2009; 37:W435-W40; PMID:19389726; http://dx.doi.org/10.1093/nar/gkp254
- Petersen T, Brunak S, Helijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrance regions. Nat Methods 2011; 8:785-6; PMID:21959131; http://dx.doi.org/10.1038/nmeth.1701
- Guo K, Li J, Tang J, Tan C, Wang H, Zeng Q. Monoclonal antibodies target intracellular PRL phosphatases to inhibit cancer metastases in mice. Cancer Biol Ther 2008; 7:750-7; PMID:18364570; http://dx.doi.org/10.4161/cbt.7.5.5764
- Hegde R, Bernstein H. The surprising complexity of signal sequences. Trends Biochem Sci 2006; 31:563-70; PMID:16919958; http://dx.doi.org/10.1016/j.tibs.2006.08.004
- Eisenhaber F, Argos P. Hydrophobic regions on protein surfaces: definition based on hydration shell structure and a quick method for their computation. Protein Eng 1996; 9:1121-33; PMID:9010925; http://dx.doi.org/10.1093/protein/9.12.1121
- Eisenhaber F. Hydrophobic regions on protein surfaces. Derivation of the solvation energy from their area distribution in crystallographic protein structures. Protein Sci 1996; 5:1676-86.
- Skjaerven L, Yao X, Scarabelli G, Grant BJ. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinformatics 2015; 15:399; ; http://dx.doi.org/10.1186/s12859-014-0399-6
- Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 2006; 22:2695-6; PMID:16940322; http://dx.doi.org/10.1093/bioinformatics/btl461
- Karagiannis P, Singer J, Hunt J, Gan S, Rudman S, Mechtcheriakova D, Knittelfelder R, Daniels T, Hobson P, Beavil A, et al. Charaterization of an engineered Trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu positive tumor cells. Cancer Immunol Immunother 2009; 58:915-30; PMID:18941743; http://dx.doi.org/10.1007/s00262-008-0607-1
- Poh J, Gan S. The determination factors involved in column-based nucleic acid extraction and purification. J Bioprocess Biotech 2014; 4:157.
- Nguyen P-V, Verma CS, Gan SK-E. DNAApp: a mobile application for sequencing data analysis. Bioinformatics 2014; 30:3270; PMID:25095882; http://dx.doi.org/10.1093/bioinformatics/btu525
- Katoh K, Standley D. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013; 30:772-80; PMID:23329690; http://dx.doi.org/10.1093/molbev/mst010
- Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res 2002; 30:E9; PMID:11788735; http://dx.doi.org/10.1093/nar/30.2.e9
- Sim JZ, Nguyen PV, Lee HK, Gan S. GelApp: mobile gel electrophoresis analyser. Nat Methods Application Notes 2015.