
ABSTRACT
Mitochondria play a key role in maintaining cellular homeostasis during stress responses, and mitochondrial dysfunction contributes to carcinogenesis, aging, and neurologic disease. We here investigated ionizing radiation (IR)-induced mitochondrial damage in human neural progenitor stem cells (NSCs), their differentiated counterparts and human normal fibroblasts. Long-term fractionated radiation (FR) with low doses of X-rays for 31 d enhanced mitochondrial activity as evident by elevated mitochondrial membrane potential (ΔΨm) and mitochondrial complex IV (cytochrome c oxidase) activity to fill the energy demands for the chronic DNA damage response in differentiated cells. Subsequent reduction of the antioxidant glutathione via continuous activation of mitochondrial oxidative phosphorylation caused oxidative stress and genomic instability in differentiated cells exposed to long-term FR. In contrast, long-term FR had no effect on the mitochondrial activity in NSCs. This cell type showed efficient DNA repair, no mitochondrial damage, and resistance to long-term FR. After high doses of acute single radiation (SR) (> 5 Gy), cell cycle arrest at the G2 phase was observed in NSCs and human fibroblasts. Under this condition, increase in mitochondria mass, mitochondrial DNA, and intracellular reactive oxygen species (ROS) levels were observed in the absence of enhanced mitochondrial activity. Consequently, cellular senescence was induced by high doses of SR in differentiated cells.
In conclusion, we demonstrated that mitochondrial radiation responses differ according to the extent of DNA damage, duration of radiation exposure, and cell differentiation.
Introduction
Mitochondria are the energy powerhouse of the cell and generate adenosine-5′-triphosphate (ATP) via oxidative phosphorylation.Citation1 IR activates mitochondrial biogenesis, which then supplies the energy required for the DNA damage response (DDR).Citation2-5 Mitochondria play a key role in maintaining the homeostasis of cells in response to IR, and function as a central platform for cellular signaling.Citation6 Mitochondria-mediated cellular signaling, including Mn-superoxide dismutase (MnSOD) and nuclear factor kB (NF-kB), is associated with the non-targeted effects of IR, such as the adaptive response, bystander effect, and genomic instability.Citation7,8 Upon high dose of IR, mitochondria release cytochrome c to induce apoptosis in irradiated cells.Citation9 Thus, mitochondria control both the survival and death of cells after IR.Citation10 We recently determined that repeated exposure to fractionated radiation (FR) with low doses of X-rays for 31 d (long-term FR) induces chronic oxidative stress in human fibroblasts via the mitochondria-mediated perturbation of reduction/oxidation (redox) control.Citation11 Elevated levels of mitochondrial ROS disturb cell cycle signaling via protein oxidation and induce genomic instability due to nuclear cyclin D1 retention in long-term low-dose irradiated cells.Citation12 Additionally, Ca2+/calcineurin-mediated retrograde signaling from mitochondria to nucleus may cause epigenetic changes in the nuclear DNA (nDNA).Citation13 Mitochondrial ROS can damage nDNA leading to genomic instability in the cells.Citation14 Altogether, mitochondria are an important cellular component of the response to long-term low-dose FR.
Mitochondria regulate ROS generation via the release of superoxide anions during oxidative phosphorylation (OXPHOS) and control of redox balance using MnSOD and glutathione (GSH) peroxidases.Citation14 The antioxidant GSH protects cells against oxygen toxicity by scavenging ROS. However, excess mitochondrial ROS due to redox perturbation leads to oxidative insults on cellular components, such as nucleic acids, proteins, and lipids. Mitochondria harbor their own mitochondrial DNA (mtDNA), which can be directly damaged by IR along with nDNA. mtDNA is located at the inner mitochondrial membrane close to the sites of ROS production and has a high mutation rate due to its chronic exposure to mitochondrial ROS.Citation15,16 The majority of the mitochondrial genome consists of genes; thus, mtDNA mutations in irradiated cells lead to mitochondrial dysfunction and radiation toxicity. To control the quality of mitochondria, IR-induced defects in this organelle are attenuated by mitochondrial fusion, in which the contents of damaged and healthy mitochondria are mixed,Citation17,18 or by the selective degradation of mitochondria (mitophagy).Citation19,20 The E3 ubiquitin ligase parkin recognizes abnormal mitochondria with low mitochondrial membrane potential (ΔΨm) and promotes their clearance via mitophagy.Citation21,22 Aberrant mitochondria cause harm to the cell. Mitochondrial functional loss alters cellular metabolism and is strongly correlated with carcinogenesis, aging, and neurodegeneration. However, the mechanism of radiation toxicity on mitochondria remains unclear.
Here we investigated IR-induced mitochondrial damage in neural progenitor stem cells (NSCs), their differentiated counterparts, and normal human fibroblasts. We observed that the radiation sensitivity of mitochondria influences the fate of irradiated cells.
Results
Mitochondrial mass in NSCs and NCs exposed to acute (SR) or long-term FR
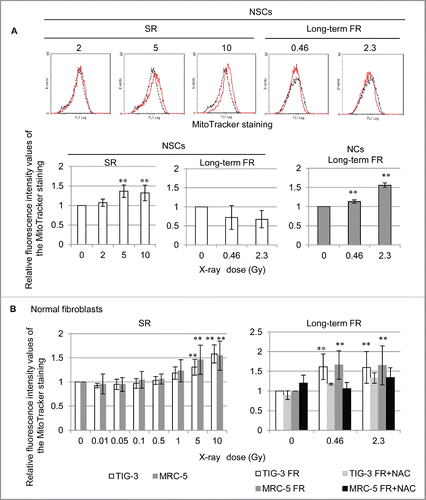
We investigated the effect of acute single radiation (SR) at range of doses between 0 and 10 Gy or long-term FR with 0.01 and 0.05 Gy/fraction for 31 d (total doses of 0.46 and 2.3 Gy, respectively) on mitochondrial mass in NSCs. MitoTracker Green FM staining revealed that the fluorescence intensity in mitochondria of NSCs was significantly increased 24 hours after >5 Gy of SR as compared with that in unirradiated NSCs whereas long-term FR had no effect (). After removing bFGF and EGF from the culturing medium, NSCs differentiated into GFAP-positive astrocytes (supplemental Fig. 1). In contrast to NSCs, these differentiated neural cells (NCs) showed increased mitochondrial mass after long-term FR ( lower right panel). We also examined mitochondrial radiation responses in other differentiated cells (i.e., TIG-3 and MRC-5 cells). High doses of SR (>5 Gy) or long-term FR increased mitochondrial mass in TIG-3 and MRC-5 cells (). Cells were continuously treated with N-acetyl-cysteine (NAC) at a final concentration of 1 mM during FR. Medium changes were made at 2- or 3-day intervals. Continuous NAC treatment eliminated the increase in mitochondria triggered by long-term FR in TIG-3 and MRC-5 cells ( right panel). These results indicate that ROS are involved in the long-term FR-induced mitochondrial mass increase in TIG-3 and MRC-5 cells.
Figure 1. Mitochondria numbers increase in response to acute SR or long-term FR. (A) Fluorescence-activated cell sorting (FACS) results for MitoTracker Green FM staining in unirradiated (dotted lines) and irradiated NSCs and NCs (solid lines). The mean fluorescence intensity values relative to unirradiated control cells are shown. (B) FACS results for MitoTracker Green FM staining in TIG-3 (white bar) and MRC-5 (gray bar) 24 hours after SR at the indicated doses shown on the left panels. The right panel shows the FACS results for MitoTracker Green FM staining in untreated 0FR (white bar), 31FR (gray bar), 0FR treated with NAC (dark gray bar), and 31FR treated with NAC cells (black bar).
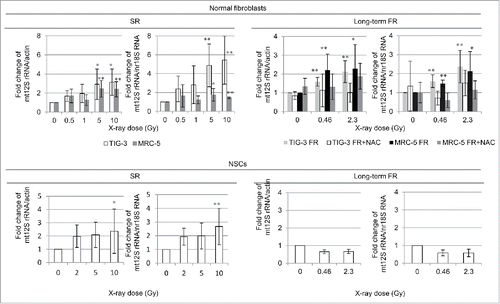
We assessed mtDNA by measuring the mtDNA copy number relative to nDNA after SR or long-term FR with quantitative PCR of 12S rRNA for detection of mtDNA and actin or 18S rRNA for detection of nDNA (). High doses of SR increased mtDNA in all cell lines as compared with that in unirradiated cells (, left panel). Long-term FR increased the mtDNA copy number in TIG-3 and MRC-5 cells but not in NSCs (, right panel). These trends are consistent with the results of the MitoTracker staining in .
Figure 2. mtDNA copy number increases after SR or long-term FR. Fold changes of mtDNA copy number relative to nDNA induced by SR or long-term FR in TIG-3, MRC-5 (upper graph), and NSCs (lower graph).
Long-term FR enhanced mitochondrial oxidative phosphorylation activity in differentiated cells but not in NSCs
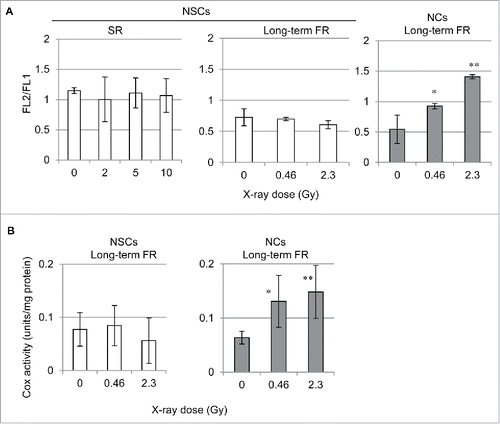
IR-induced increases in mitochondrial mass may be due to enhanced mitochondrial activity in irradiated cells. Therefore, we examined the radiation effects on mitochondrial function in human irradiated cells. The mitochondrial membrane potential (ΔΨm) in NSCs and NCs was measured by staining with the lipophilic cation JC-1.Citation23 As expected, long-term FR elevated the ΔΨm in NCs ( right panel). In contrast, SR or long-term FR did not affect the ΔΨm in NSCs ( center and left panels). We examined mitochondrial complex IV (cytochrome c oxidase (COX)) activity to investigate mitochondrial OXPHOS. Consistent with the elevated ΔΨm, long-term FR enhanced COX activity in NCs ( right panel). In addition to the lack of effects on the number of mitochondria and ΔΨm, COX activity remained unchanged after long-term FR in NSCs ( left panel).
Figure 3. Mitochondrial membrane potential and COX activity after IR. (A) FACS results for JC-1 staining after SR or long-term FR in NSCs and NCs. (B) Cytochrome c oxidase (COX) activity after long-term FR in NSCs and NCs.
Efficient DNA repair of long-term FR-induced nuclear DNA damage in NSCs
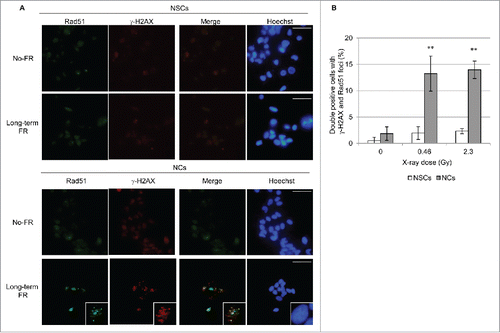
Long-term FR-induced mitochondrial activation is associated with the energy demands for double-strand breaks (DSBs) repair. We investigated DSBs repair by immunostaining with γ-H2AX, the marker for DSBs, and Rad51, a marker for homologous recombination repair, in NSCs and NCs. Long-term FR induced prolonged DSBs repair as evidenced by the formation of γ-H2AX and RAD51 foci in NCs 24 hours after the last FR exposure ( and ). In contrast, NSCs showed no γ-H2AX and RAD51 foci after long-term FR (and ). Thus, NSCs resisted long-term FR-induced damage through the efficient DNA repair of DSBs.
Figure 4. γ-H2AX and Rad51 foci formation after long-term FR. (A) Images of γ-H2AX (red) and Rad51 (green) positive cells in NSCs and NCs. DNA was stained with Hoechst. Magnified images of a particular cell are shown in the insert. The scale bar represents 50 µm. (B) The percentage of γ-H2AX and Rad51 double-positive NSCs and NCs.
Mitochondrial ROS and oxidative damage in response to SR and long-term FR
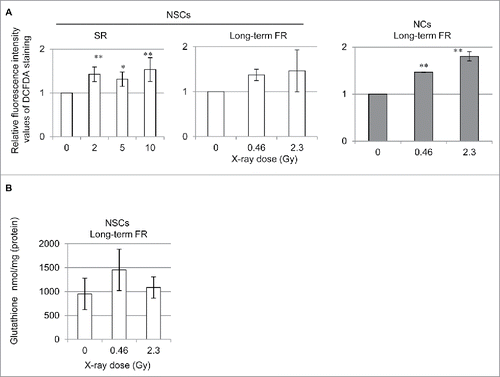
ROS are released from mitochondria as a byproduct of OXPHOS.Citation14 Mitochondrial ROS levels were investigated with 2′,7′-dichlorofluorescin diacetate (DCFDA) staining in cells 24 hours after IR (). As previously reported in TIG-3 and MRC-5 cells,Citation11 ROS levels increased by long-term FR in NCs (right panel). Long-term FR did not affect ROS or GSH levels in NSCs since there was no induction of mitochondrial OXPHOS (). In contrast, SR caused an increase in ROS levels as compared with those in the unirradiated NSCs in the absence of elevated ΔΨm (left panels).
Figure 5. ROS generation in 0FR and 31FR cells. (A) FACS results for DCFDA staining after SR in NSCs and NCs. The relative fluorescence intensity values of DCFDA staining normalized to unirradiated controls are shown. (B) GSH levels in NSCs after long-term FR.
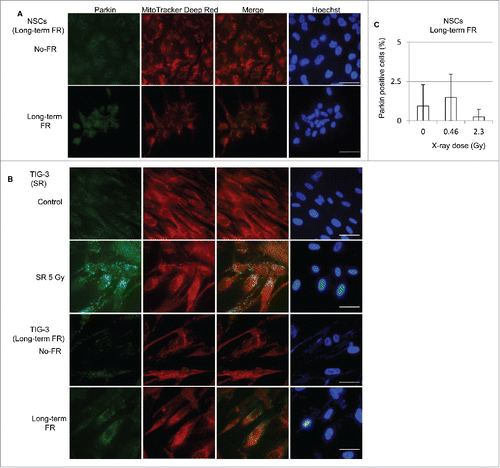
To identify ROS-mediated mitochondrial damage, we immunostained for the E3 ubiquitin ligase parkin, which marks mitochondria with low ΔΨm for degradation via mitophagy. The mitochondrial damage detected by parkin staining was not induced by long-term FR in NSCs consistent with no induction of ROS (). As previously reported, long-term FR induced parkin-positive cells in TIG-3 and MRC-5 cells ().Citation24 Parkin also localized to damaged mitochondria after 5 Gy of SR in TIG-3 cells, as shown by the high intensity yellow color in the merged panel ().
Figure 6. Immunofluorescent staining for parkin. (A) Images of parkin staining in NSCs (B) Localization of parkin in unirradiated (control) TIG-3 cells and TIG-3 cells irradiated with 5 Gy. DNA was stained with Hoechst. The scale bar represents 50 µm. (B) Percentage of parkin-positive NSCs exposed to FR at doses of 0.01 or 0.05 Gy/fraction.
Cell cycle arrest and cellular senescence induced by SR at high doses
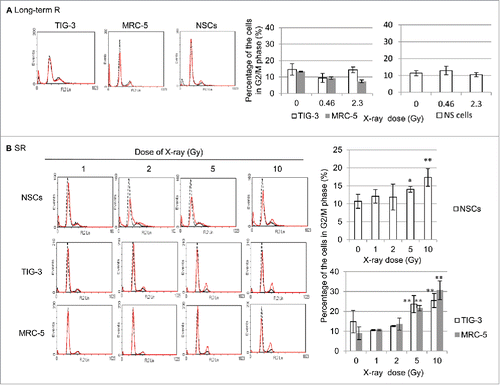
To clarify the deleterious impact of mitochondrial ROS-mediated oxidative stress in irradiated cells, the fate of cells was investigated after SR and long-term FR. The cell cycle distribution was examined by propidium iodide (PI) staining using fluorescence-activated cell sorter (FACS) in NSCs, TIG-3, MRC-5, after IR. As we previously reported that cells continued to grow exponentially for 31 d if exposed to 0.01 or 0.05 Gy FR.Citation12 The histograms of cellular DNA content indicated that long-term FR had a negligible effect on IR-induced G2-arrest in TIG-3, MRC-5, and NSCs (). In contrast, high doses of SR induced G2-phase cell cycle arrest in all cell lines ().
Figure 7. G2 cell cycle arrest after high doses of SR. (A) FACS results of PtdIns staining in unirradiated (dotted lines) and irradiated TIG-3, MRC-5, and NSCs (dot lines) after long-term FR (left panel). Percentage of cells in G2/M phase (right graph). (B) FACS results of PtdIns staining in unirradiated (dotted lines) and irradiated TIG-3, MRC-5, and NSCs (dot lines) (left panel). Percentage of cells in G2/M phase (right panel).
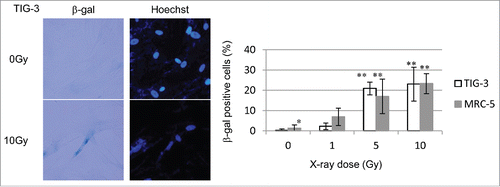
ROS generation is associated with the induction of senescence via oxidative stress. Senescent cells were identified by detection of senescence-associated β-galactosidase (β-gal) activity. shows representative β-gal staining in TIG-3 cells after 10 Gy of SR. The percentage of β-gal-positive senescent cells increased in a dose-dependent manner in MRC-5 and TIG-3 cells (). Our results indicate that high doses of SR-induced mitochondrial damage led to G2-phase cell cycle arrest and cellular senescence in irradiated cells.
Figure 8. Cellular senescence after low-dose long-term FR. (A) Images of β-gal staining in unirradiated control cells and irradiated 31FR MRC-5 cells with and without NAC treatment. Percentage of β-gal-stained cells in unirradiated control and irradiated 31FR cells with and without NAC treatment.
Discussion
Radiation dose response of mitochondria in human cells
Understanding how mitochondria respond to IR and how cellular metabolism brings about IR-induced carcinogenesis, aging, and neurologic disease is important for the identification the critical health aspects of radiation toxicity in humans. Here we demonstrated that high doses of acute SR induced an increase in mitochondrial mass in human cells, whereas less than 1 Gy of SR did not affect mitochondria. A previous study showed that mtDNA replication did not occur concurrently with nDNA replication and was not inhibited by IR.Citation25 Indeed, our data showed that high doses of SR increased the number of mitochondria during G2 arrest. Strong parkin staining indicated that mitochondria were severely injured in TIG-3 and MRC-5 cells after high doses of SR. In addition, a loss of respiratory function was detected by JC-1 staining. It was concluded that ROS was released from damaged mitochondria and this brought about cellular senescence in irradiated TIG-3 and MRC-5 cells.
Resistance to long-term FR in NSCs
The DDR varies according to the radiation dose and the duration of radiation exposure.Citation26 Here we demonstrated that long-term FR induced prolonged DSBs repair, as evidenced by the persistent γ-H2AX and RAD51 foci in NCs. We previously reported that long-term FR upregulated the expression of PPAR-γ co-activator-1 α (PGC1-α), which stimulates mitochondrial biogenesis.Citation11 Mitochondria are necessary for some of the processes in the DDR due to their energy requirements, including DNA repair, cell cycle checkpoints, and apoptosis. Long-term FR induced a high ΔΨm and enhanced COX activity, which highlights the association between nDNA damage and the mitochondrial energy supply for adaptive responses against long-term FR. We previously reported that enhanced mitochondrial activity resulted in perturbation of redox control, which then abrogated AKT/cyclin D1 cell cycle signaling via oxidative inactivation of protein phosphatase 2A. The resulting abnormal nuclear retention of cyclin D1 induced genomic instability in long-term FR irradiated cells.Citation10-12 In contrast, NSCs showed the lack of effects on induction of mitochondrial activity by long-term FR. These cells exhibited efficient DNA repair during FR exposure intervals and resistance to long-term FR as evident by no γ-H2AX and RAD51 foci. Several reports demonstrated an important role for mitochondria in the differentiation of stem cells.Citation27 The metabolic transition from glycolysis to OXPHOS in mitochondria is necessary for cell differentiation. Mitochondria undergo significant changes to provide higher amounts of energy to sustain specialized functions in different tissues. When NSCs were differentiated in to NCs, long-term FR enhanced mitochondrial ΔΨm and COX activity. This continuous mitochondrial activation in NCs resulted in mitochondrial oxidative stress via ROS.
Mitochondrial dysfunction associated with radiation-induced brain damage
Tumors receive a large total radiation dose via FR during radiation therapy (RT). The surrounding off-target normal brain tissue also receives low doses of IR during RT for brain tumors. Therefore, therapeutic irradiation of the brain can lead to harmful radiation effects, including cognitive impairment. In vitro approaches to study neural precursor cell radiation responses will help provide appropriate solutions to this problem. Here we demonstrated that differentiated NCs were more susceptible to long-term FR than the parental NSCs. Mitigation of radiation-induced brain injury is a critical issue in the development of better approaches for cancer treatment in the brain. Protection against mitochondrial oxidative damage by treatment with antioxidants is useful to suppress the radiation toxicity of neural cells during fractionated RT. NAC serves as a cysteine donor for the synthesis of GSHCitation28 and increases intracellular levels of GSH for ROS scavenging.
In conclusion, we demonstrated differences in the mitochondrial radiation response to long-term FR between NSCs and differentiated cells. Mitochondria engaged for the cellular response to long-term FR and determine the fate of irradiated cells. Mitochondrial activation induces oxidative stress after long-term FR. The subsequent oxidative damage to mtDNA accumulates and leads to mutagenesis, carcinogenesis, accelerated senescence, and cell death. Therefore, antioxidants are useful agents for protection against the mitochondrial damage induced by long-term FR.
Materials and methods
Cell culture conditions and drugs
Normal human diploid lung fibroblasts (MRC-5 and TIG-3), neural progenitor stem cells (NSCs: immortalized ReNcell VM Human Neural Progenitor Cell line) and differentiated neural cells (NCs) were used. MRC-5 and TIG-3 were purchased from the Health Science Research Resources Bank and grown in minimum essential medium (Nacalai Tesque) supplemented with 10% heat-inactivated fetal calf serum. The NSCs derived from a 10-week-old human fetus was purchased from Milipore and grown in ReNcell NSC Maintenance Medium containing basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF) according to the manufacturer's protocol. For the initiation of differentiation, cells were grown in ReNcell NSC Maintenance Medium without bFGF and EGF to generate NCs. NAC was purchased from Sigma.
Irradiation experiments
Cells were irradiated using a 150-kVp X-ray generator (Model MBR-1505R2, Hitachi) with 0.5-mm Cu and 0.1-mm Al filters. The dose rates were 0.11 Gy/min for FR with 0.01Gy, and 0.49 Gy/min for FR with 0.05Gy or SR. Low dose X-ray fractions (0.01 or 0.05 Gy) were administrated twice a day, 5 days/week. The total doses delivered over 31 d were 0.46 Gy and 2.3 Gy for cells exposed to FR of 0.01 Gy and 0.05 Gy, respectively. Cells continued to grow for 31 d if exposed to 0.01 or 0.05 Gy FR. When cells reached 80% of confluency, cells were subcultured in a new flask.
Mitochondrial mass and ROS measurements
Cells were stained with 20-µM DCFDA (Sigma) or 400-nM MitoTracker Green FM (Invitrogen, Carlsbad) in minimum essential medium without serum for 30 min 24 h after SR or the last FR. DCFDA- or MitoTracker Green FM-stained cells were quantified with FACScan (Becton Dickinson). Cells were placed on glass slides and cultured overnight. Cells on coverslips were stained with MitoTracker Green FM according to the manufacturer's instructions (Invitrogen). Images were acquired using a CCD camera attached to a fluorescence microscope (Keyence).
Quantification of mtDNA copy number
Quantitative real-time polymerase chain reaction (QPCR) was performed using SUBR Green PCR Master Mix (Applied Biosystems). A mitochondrially encoded 12S rRNA (forward primer 5′-AGAACACTACGAGCCACAGC -3′, reverse primer 5′-ACTTGCGCTTACTTTGTAGCC-3′) was used for the mitochondrial genome. 18S rRNA (forward primer 5′-GGAGTATGGTTGCAAAGCTG-3′, reverse primer 5′-CGCTCCACCAACTAAGAACG-3′) and GAPDH (forward primer 5′-TACTGGTGTCTTCACCACCA-3′, reverse primer 5′-CAGGATGCATTGCTGACAATC-3′) were used for the nuclear genome. Total cell DNA was extracted with a DNA extraction WB kit (Wako). PCR reactions were performed for 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s using a Stratagene Mx3000P QPCR system (Agilent Technologies Inc.). All samples were assayed in triplicate. The relative mtDNA/nDNA ratio was defined as the mitochondrial copy number and determined using the comparative threshold cycle (CT) method.
Mitochondrial membrane potential
Cells were stained with JC-1 (tetraethylbenzimidazolylcarbocyanine iodide) according to the manufacturer's instructions (Invitrogen). Cell s were incubated in JC-1 for 15 min. JC-1 stained cells were quantified with FACScan (Becton Dickinson).
Measurement of cytochrome c oxidase activity
COX activity was measured using a cytochrome c oxidase assay kit (Sigma) according to the manufacturer's instructions. The activity was reported as units/mg of cell extract.
Immunofluorescence
Immunofluorescence staining was performed as described previously.Citation12,29 Cells were fixed with 4% formaldehyde for 10 min and permeabilized with 0.5% Triton X-100 for 5 min. For double staining with MitoTraker deep red (Invitrogen) and parkin, cells were fixed with 4% formaldehyde for 15 min and permeabilized with 0.2% Triton X-100 for 10 min. Antibodies against γ-H2AX (Millipore, Billerica, MA, USA), Parkin (Santa Cruz Biotechnology), and rad51 (Nichirei Bioscience) and secondary antibodies conjugated with Alexa Fluor 488 (Molecular Probes, Eugene, OR, USA) or Cy-3 (Jackson ImmunoResearch Laboratories) were used. Cells were counterstained for DNA with Hoechst 33258 (4 µg/mL in Vectashield mounting medium; Vector Laboratories). Images were captured using a CCD camera attached to a fluorescence microscope (Keyence). For each data point, >50 cells were counted from at least 3 independent samples.
Measurement of glutathione
Total Glutathione was quantified using a Total Glutathione Quantification Kit (Dojindo) according to the manufacturer's protocol. OD values at 405 nm were measured with a Sunrise microplate reader (Tecan).
Cell cycle analysis
Cells were fixed in 70% ethanol at −20°C for >30 min. DNA was stained with PtdIns in the presence of RNase. PtdIns-positive cells were detected and quantified by FACScan (Becton Dickinson).
Senescence
Cellular senescence was examined using a Senescence Detection Kit (BioVision) according to the manufacturer's instructions. Images were captured using a CCD camera attached to a fluorescence microscope (Keyence). For each data point >50 cells were counted from at least 3 independent samples.
Statistical analysis
Error bars represent the standard deviation. All experiments were repeated at least 3 times using independent samples. Dunnett test following one-way ANOVA was used to detect significant differences between the means of 3 or more independent groups. Double and single asterisks indicate significant differences with p-values of <0.01 and <0.05, respectively.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1284716_Supplemental_Material.zip
Download Zip (189 KB)Acknowledgments
This work was performed at the Joint Usage/Research Center (Radiation Biology Center), Kyoto University and the Program of the network-type joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University and Fukushima Medical University. The authors would like to thank Enago (www.enago.jp) for the English language review.
Funding
Sources of support: This research was supported by a grant from the Japanese Ministry of Education and Science Houga (15K12220, 15K12212), Industrial Disease Clinical Research Grants from the Japanese Ministry of Health, Labor, and Welfare and in part by NIFS Collaborative Research Program (NIFS13KOBA028).
Related Research Data
References
- Ernster L, Schatz G. Mitochondria: a historical review. J Cell Biol 1981; 91:227s-55s; PMID:7033239; http://dx.doi.org/10.1083/jcb.91.3.227s
- Yu J, Wang Q, Chen N, Sun Y, Wang X, Wu L, Chen S, Yuan H, Xu A, Wang J. Mitochondrial transcription factor A regulated ionizing radiation-induced mitochondrial biogenesis in human lung adenocarcinoma A549 cells. J RadiatRes 2013; 54:998-1004; PMID:23645454; http://dx.doi.org/10.1093/jrr/rrt046
- Kulkarni R, Marples B, Balasubramaniam M, Thomas RA, Tucker JD. Mitochondrial gene expression changes in normal and mitochondrial mutant cells after exposure to ionizing radiation. Radiat Res 2010; 173:635-44; PMID:20426663; http://dx.doi.org/10.1667/RR1737.1
- Gong B, Chen Q, Almasan A. Ionizing radiation stimulates mitochondrial gene expression and activity. Radiat Res 1998; 150:505-12; PMID:9806591; http://dx.doi.org/10.2307/3579866
- Yamamori T, Yasui H, Yamazumi M, Wada Y, Nakamura Y, Nakamura H, Inanami O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radical Biol Med 2012; 53:260-70; http://dx.doi.org/10.1016/j.freeradbiomed.2012.04.033
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem 2012; 287:4434-40; PMID:21832045; http://dx.doi.org/10.1074/jbc.R111.271999
- Kim GJ, Fiskum GM, Morgan WF. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res 2006; 66:10377-83; PMID:17079457; http://dx.doi.org/10.1158/0008-5472.CAN-05-3036
- Eldridge A, Fan M, Woloschak G, Grdina DJ, Chromy BA, Li JJ. Manganese superoxide dismutase interacts with a large scale of cellular and mitochondrial proteins in low-dose radiation-induced adaptive radioprotection. Free Radical Biol Med 2012; 53:1838-47; http://dx.doi.org/10.1016/j.freeradbiomed.2012.08.589
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15:2922-33
- Shimura T, Kunugita N. Mitochondrial reactive oxygen species-mediated genomic instability in low-dose irradiated human cells through nuclear retention of cyclin D1. Cell Cycle 2016; 15:1410-4; PMID:27078622; http://dx.doi.org/10.1080/15384101.2016.1170271
- Shimura T, Sasatani M, Kamiya K, Kawai H, Inaba Y, Kunugita N. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget 2016; 7:3559-70; PMID:26657292
- Shimura T, Hamada N, Sasatani M, Kamiya K, Kunugita N. Nuclear accumulation of cyclin D1 following long-term fractionated exposures to low-dose ionizing radiation in normal human diploid cells. Cell Cycle 2014; 13:1248-55; PMID:24583467; http://dx.doi.org/10.4161/cc.28139
- Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013; 13:577-91; PMID:24004957; http://dx.doi.org/10.1016/j.mito.2013.08.007
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005; 120:483-95; PMID:15734681; http://dx.doi.org/10.1016/j.cell.2005.02.001
- Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A 1988; 85:6465-7; PMID:3413108; http://dx.doi.org/10.1073/pnas.85.17.6465
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 1997; 94:514-9; PMID:9012815; http://dx.doi.org/10.1073/pnas.94.2.514
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007; 130:548-62; PMID:17693261; http://dx.doi.org/10.1016/j.cell.2007.06.026
- Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genetics 2001; 28:272-5; PMID:11431699; http://dx.doi.org/10.1038/90116
- Tolkovsky AM. Mitophagy. Biochim Et Biophys Acta 2009; 1793:1508-15; PMID:19289147; http://dx.doi.org/10.1016/j.bbamcr.2009.03.002
- Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circulation Res 2012; 111:1208-21; PMID:23065344; http://dx.doi.org/10.1161/CIRCRESAHA.112.265819
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 2003; 100:4078-83; PMID:12642658; http://dx.doi.org/10.1073/pnas.0737556100
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795-803; PMID:19029340; http://dx.doi.org/10.1083/jcb.200809125
- Perelman A, Wachtel C, Cohen M, Haupt S, Shapiro H, Tzur A. JC-1: alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis 2012; 3:e430; http://dx.doi.org/10.1038/cddis.2012.171
- Shimura T, Kobayashi J, Komatsu K, Kunugita N. Severe mitochondrial damage associated with low-dose radiation sensitivity in ATM- and NBS1-deficient cells. Cell Cycle 2016; 15:1099-107; PMID:26940879; http://dx.doi.org/10.1080/15384101.2016.1156276
- Cleaver JE. Replication of nuclear and mitochondrial DNA in X-ray-damaged cells: evidence for a nuclear-specific mechanism that down-regulates replication. Radiat Res 1992; 131:338-44; PMID:1438691; http://dx.doi.org/10.2307/3578425
- Shimura T, Fukumoto M, Kunugita N. The role of cyclin D1 in response to long-term exposure to ionizing radiation. Cell Cycle 2013; 12:2738-43; PMID:23974042; http://dx.doi.org/10.4161/cc.25746
- Xu X, Duan S, Yi F, Ocampo A, Liu GH, Izpisua Belmonte JC. Mitochondrial regulation in pluripotent stem cells. Cell Metab 2013; 18:325-32; PMID:23850316; http://dx.doi.org/10.1016/j.cmet.2013.06.005
- Staal FJ, Roederer M, Herzenberg LA, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci U S A 1990; 87:9943-7; PMID:2263644; http://dx.doi.org/10.1073/pnas.87.24.9943
- Shimura T, Toyoshima M, Adiga SK, Kunoh T, Nagai H, Shimizu N, Inoue M, Niwa O. Suppression of replication fork progression in low-dose-specific p53-dependent S-phase DNA damage checkpoint. Oncogene 2006; 25:5921-32; PMID:16682953; http://dx.doi.org/10.1038/sj.onc.1209624