
Mechanistic target of rapamycin complex 1 (mTORC1) lies at the center of nutrient sensing, and is involved in numerous pathological and physiologic processes. mTORC1 receives a wide variety of upstream inputs and in turn controls an equally large number of downstream biologic events through phosphorylation of its substrates. Largely unanswered, however, is how and when substrate specificity of mTORC1 is regulated in response to different upstream inputs. Our recent work on the role of mTORC1 in adipose tissue browning is now providing some insight into this question.
Browning of adipose tissue is a biologic process where white adipose tissue (WAT) acquires thermogenic metabolism in response to cold challenge through accelerated mitochondrial biogenesis. Published studies on the role of mTORC1 in the context of adipose tissue browning have been paradoxical. Hall's groupCitation1 generated fat-specific knockouts (adipKO) of raptor, an essential component of mTORC1, and showed that the mice were resistant to obesity upon high fat diet challenge, had increased systemic respiration, and demonstrated browning of WAT. These data thus suggested that mTORC1 suppresses browning. Recently, however, Baur's group and Collins' group independently reported that mTORC1 plays an essential role in cold-inducible browning of WAT.Citation2,3 The groups inhibited mTORC1 pharmacologically with systemic administration of rapamycin, or genetically with fat-specific raptor deletion, and showed that either approach ablates browning of WAT upon cold exposure. Mechanistically, protein kinase A (PKA) directly phosphorylates raptor upon β adrenergic receptor (βAR) stimulation, leading to phosphorylation of the canonical mTORC1 substrate S6K, and subsequent induction of a downstream browning program.Citation3 Interestingly, in this study too, raptor adipKO animals also showed mild browning phenotype before cold challenge. Together, these data suggest the somewhat paradoxical conclusion that mTORC1 stimulates browning in response to cold exposure or βAR stimulation, but that mTORC1 inhibits browning at thermoneutrality.
Our recent work may now reconcile this paradox, in the context of mTORC1 substrate specificity. We have identified a novel mTORC1-TFE3 pathway that regulates adipose tissue browning downstream of FLCN.Citation4 FLCN was originally identified as mutated in patients with the BHD syndrome, a hamartomatous disease marked by renal cancers with hyperactive mitochondrial biogenesis. How does FLCN suppress mitochondrial biogenesis? We identified the bHLH transcription factor TFE3 as a key factor downstream of FLCN. Upon loss of FLCN, TFE3 localizes to nucleus, where TFE3 induces a browning program via direct activation of the potent transcriptional coactivator PGC-1β. How does FLCN suppress TFE3 nuclear localization? Interestingly, mTOR inhibition by torin1 also sends TFE3 to nucleus. It has been reported that TFEB, a transcription factor highly homologous to TFE3, is also phosphorylated by mTORC15, leading to 14–3–3 binding and sequestration in the cytoplasm.Citation5 Based on the amino acid sequence similarity between these factors, we identified Ser 320 residue of mouse TFE3 (Ser 321 in human TFE3) as an mTORC1 phosphorylation site. FLCN deletion or pharmacological mTOR inhibition blunts TFE3 phosphorylation at Ser 320, and alanine substitution of the Ser 320 constitutively sends TFE3 to nucleus. In vivo, adipose-specific loss of FLCN leads to adipose browning, which is entirely rescued by co-deletion of either TFE3 or PGC-1β. These results show that FLCN inhibits TFE3 localization through mTORC1, and that TFE3 in turn regulates adipose tissue browning.
The surprise in our results came when we evaluated the effect of FLCN deletion on other branches downstream of mTORC1, notably phosphorylation of S6K. S6K phosphorylation remained intact in FLCN deficient adipocytes, in sharp contrast to TFE3 phosphorylation. Conversely, hyper-activation of mTORC1 by disruption of the inhibitory Tsc1/2 complex, led to hyper-phosphorylation of S6K but failed to prevent TFE3 nuclear localization upon FLCN deletion. Together, our data show that the mTORC1-TFE3 branch is separable from the canonical mTORC1-S6K branch, and FLCN regulates the former branch but not the latter. In light of these results, we can now reconcile the paradox regarding contradictory mTORC1 functions in adipose tissue browning. Cold induced browning occurs via PKA-mediated activation of the canonical mTORC1-S6K branch, while baseline browning is suppressed by the mTORC1-TFE3 branch. Loss-of-raptor blunts both branches of mTORC1, thus constitutively sending TFE3 to the nucleus to induce browning at baseline, while at the same time βAR-PKA signals fail to induce browning program upon cold challenge ().
Figure 1. Model of 2 separable mTORC1 branches regulating adipose tissue browning. Cold stimulation/β-adrenergic signaling activates mTORC1 through PKA to induce browning. On the other hand, FLCN suppresses TFE3 nuclear localization via mTORC1 through control of RagC/D guanyl nucleotide charging status, which is separable from the mTORC1-S6K branch. Loss-of-mTORC1 ablates browning response upon cold stimulation while, at the same time, leads to constitutive hyper-activation of TFE3, which induces browning at baseline.
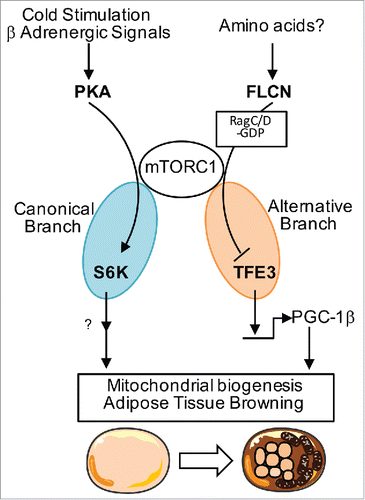
Numerous questions remain. How does FLCN regulate mTORC1 activity in a branch/substrate-specific manner? Previously, FLCN was identified as GTPase activating protein (GAP) for RagC/DCitation6, which heterodimerizes with RagA/B to signal amino acid level to mTORC1.Citation7 We showed that constitutively active, GDP-bound Rag C/D sends TFE3 back to the cytoplasm in FLCN KO cells. These data imply that RagC/D is mostly GTP bound without FLCN, and fails to recruit TFE3 to lysosome surface. It is thus possible that the guanyl nucleotide charging status of RagC/D could define substrate specificity of mTORC1 by recruiting a subset of mTOR substrates to the lysosome surface, a site where mTORC1 is known to be activated. Another critical question is what signals act upstream of FLCN? We showed that amino acid stimulation localizes TFE3 to the cytoplasm, but cells lacking FLCN failed to do so, suggesting a key role in amino acid sensing. It remains unclear which specific amino acid(s) are involved, or how mechanistically the process occurs.
It remains a daunting challenge to understand how specific inputs to mTORC1 are linked to specific corresponding outputs of mTORC1 action. Our study provides a clear example of such specific branching, with relevance to systemic physiology. If mTORC1 inhibition is ultimately to have specific clinical applications, it will likely need to similarly branch out.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Polak P, Cybulski N, Feige JN, Auwerx J, Rüegg MA, Hall MN. Adipose-Specific Knockout of raptor Results in Lean Mice with Enhanced Mitochondrial Respiration. Cell Metab 2008; 8:399-410; PMID: 19046571; http://dx.doi.org/10.1016/j.cmet.2008.09.003
- Tran CM, Mukherjee S, Ye L, Frederick DW, Kissig M, Davis JG, Lamming DW, Seale P, Baur JA. Rapamycin blocks induction of the thermogenic program in white adipose tissue. Diabetes 2016; 6060:1-35; http://dx.doi.org/10.2337/db15-0502
- Liu D, Bordicchia M, Zhang C, Fang H, Wei W, Li J, Guilherme A, Guntur K, Czech MP, Collins S. Activation of mTORC1 is essential for β-adrenergic stimulation of adipose browning. J Clin Invest 2016; 126:1704-16; PMID: 27018708; http://dx.doi.org/10.1172/JCI83532
- Wada S, Neinast M, Jang C, Ibrahim YH, Lee G, Babu A, Li J, Hoshino A, Rowe GC, Rhee J, et al. The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev 2016:1-14; PMID: 26728553; http://dx.doi.org/10.1101/gad.287953.116
- Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012; 8:903-14; PMID: 22576015; http://dx.doi.org/10.4161/auto.19653
- Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, Spooner E, Sabatini D. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 2013; 52:495-505; PMID: 24095279; http://dx.doi.org/10.1016/j.molcel.2013.09.016
- Sancak Y, Peterson TR, Shaul YD, Lindquist R a, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320:1496-501; PMID: 18497260; http://dx.doi.org/10.1126/science.1157535