
ABSTRACT
Genomic instability is a hallmark of cancer and a common feature of human disorders, characterized by growth defects, neurodegeneration, cancer predisposition, and aging. Recent evidence has shown that DNA replication stress is a major driver of genomic instability and tumorigenesis. Cells can undergo mitosis with under-replicated DNA or unresolved DNA structures, and specific pathways are dedicated to resolving these structures during mitosis, suggesting that mitotic rescue from replication stress (MRRS) is a key process influencing genome stability and cellular homeostasis. Deregulation of MRRS following oncogene activation or loss-of-function of caretaker genes may be the cause of chromosomal aberrations that promote cancer initiation and progression. In this review, we discuss the causes and consequences of replication stress, focusing on its persistence in mitosis as well as the mechanisms and factors involved in its resolution, and the potential impact of incomplete replication or aberrant MRRS on tumorigenesis, aging and disease.
Introduction
Genomic stability relies on accurate and complete genome duplication and faithful chromosome segregation during cell division. Cells are continuously exposed to exogenous or endogenous insults that may lead to DNA damage and challenge DNA replication. The eukaryotic cell has devised different mechanisms, known as DNA damage response (DDR), to detect, signal and repair DNA damage. The DDR involves a complex signaling cascade mediated by phosphatidylinositol 3-kinase-related kinases (PIKKs), including DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia-mutated (ATM), and ATM and Rad3-related (ATR) proteins. These sensor kinases phosphorylate a plethora of substrates, including DNA repair factors and checkpoint proteins, to coordinate DNA repair and chromatin modifications with cell cycle progression and cellular metabolic processes.Citation1 In addition to damaged templates, replication fork progression can be challenged by intrinsic obstacles such as difficult-to-replicate loci, highly transcribed genes, repeat sequences, non-canonical DNA structures or protein-DNA adducts.Citation2 All conditions that perturb DNA replication leading to replication fork stalling or slowing are collectively referred to as DNA replication stress. Replication stress induces a specialized branch of the DDR, the replication or S-phase checkpoint, triggered by the generation of stretches of single-stranded DNA at stalled or damaged forks, and depends on the activation of ATR and Chk1 checkpoint kinases.Citation3-5 This pathway is fundamental to delaying cell cycle progression and allowing the cell to properly and timely repair damage, recover stalled replication forks, and complete replication before entry into mitosis. However, endogenous or low levels of replication stress or DNA damage, might not be sensed by the cell and escape checkpoint activation. Moreover, completion of DNA replication at certain genomic regions can be delayed up to G2 phase or even mitosis, leading to persistence of under-replicated DNA in mitosis, especially in replication stress conditions.Citation6-8
Mitosis is the complex cellular process by which duplicated genetic material is equally distributed to 2 daughter cells.Citation9 It begins with chromosome condensation (prophase), centrosome separation to form a bipolar spindle and attachment of the kinetochores to the spindle microtubules (prometaphase). By metaphase, all chromosomes become aligned on the metaphase plate and bioriented with respect to the spindle poles, which is essential for faithful segregation of the chromosomes into the daughter cells. Then, anaphase occurs, characterized by the separation and movement of sister chromatids toward the 2 opposite poles of the spindle. Mitosis ends with telophase, when the 2 sets of chromosomes reach the spindle poles and chromatin decondenses before cytokinesis physically separates the 2 daughter cells.
Mitotic defects leading to unequal chromosome segregation are a common cause of chromosomal instability (CIN) and cancer.Citation10 Defective chromosome segregation and aneuploidy can be caused by dysfunction of the factors regulating proper assembly and dynamics of the mitotic apparatus, sister chromatid cohesion, bipolar attachment of kinetochores to the mitotic spindle and the spindle assembly checkpoint.Citation11 However, faithful chromosome segregation and maintenance of genome stability also depend on the coordination between genome duplication and cell cycle and mitotic progression. Recent studies have shown that under-replicated or unresolved DNA structures that form as a consequence of replication stress and persist into mitosis may hamper chromosome segregation leading to micronucleation and gain or loss of genetic material.Citation12-14 They have highlighted the existence of pathways that operate not only during S-phase but also during mitosis to limit the deleterious consequences of replication stress and the transmission of DNA damage to daughter cells.Citation15 Works from several groups have started to identify some of the factors and mechanisms involved.Citation12, 13, 16-24 Collectively, they suggest that mitotic rescue from replication stress (MRRS) is a key process that must be tightly regulated to prevent structural and numerical chromosomal aberrations and avoid mitotic catastrophe and other deleterious outcomes of defective chromosome segregation. Therefore, we propose that exploring how cells respond to replication stress during mitosis may be crucial to understanding the mechanisms of replication stress-driven genomic instability. In this review, we discuss the causes of replication stress and the consequences of unresolved DNA damage or incomplete DNA replication, specifically in mitosis. We also present recent advances in the knowledge of the cellular response to under-replicated DNA in mitosis, with a specific focus on common fragile sites (CFSs). Finally, we discuss the different pathways involved in the rescue of DNA damage in mitosis and the consequences of their dysfunction on human health.
DNA replication program and the causes of replication stress
DNA replication is initiated at specific genomic loci called origins of replication.Citation25 It occurs bidirectionally and proceeds through the regulated and sequential firing of replication origins, leading to faithful and complete duplication of the chromosomes. Replication stress can be the outcome of a low density or inefficient activation of origins leading to long traveling and unstable replication forks, which makes origin-poor regions particularly sensitive to replication stress.Citation26-29 Alternatively, replication stress can be the result of firing of an excessive number of origins, as a consequence of a defect in the developmental program of replication or the cellular replication timing program.Citation30 In such conditions, factors required for DNA replication, such as replication initiation factors, deoxyribonucleotides (dNTPs), the single-strand DNA binding protein RPA (replication protein A) and histones may become limiting, causing elongation problems and subsequent fork collapse.Citation31, 32 The number of replication origins that are loaded (licensed) in G1 by the replicative MCM2–7 helicase is in excess with respect to the origins that are fired during a normal cell cycle.Citation33 A large fraction of them are passively replicated by forks coming from nearby origins and remain dormant in unperturbed conditions. However, they serve as back-up origins in case of replication fork stalling or slowing and become essential to ensure complete replication in replicative stress conditions.Citation27, 34-36 If a replication fork stalls and DNA synthesis does not readily resume, stabilization of the arrested fork, which allows DNA to be replicated from an incoming adjacent fork, as well as firing of additional origins locally to compensate for replication slowdown are both important mechanisms by which cells respond or adapt to replication stress.Citation37-39 If stalling occurs at converging forks in a region lacking replication origins or at unidirectional forks, cells must rescue the fork. ATR, the central kinase involved in the replication stress response, not only delays cell cycle progression but also regulates origin firing and orchestrates many different pathways involved in fork stabilization, remodeling, and restart or repair of stalled or collapsed forks.Citation4,40,41 The primary pathways that allow resumption of DNA synthesis in the event of replication obstacles include fork repriming, translesion synthesis or lesion bypass by template switching, fork remodeling/processing and fork restart by homologous recombination-mediated mechanisms.Citation42 The choice of the pathway used to restart replication forks is a matter of intense investigation and likely depends on the type of the blocking lesion or DNA structure, the genomic or chromatin context, and the timing at which replication stalling occurs.
The different causes of replication stress have been recently reviewed.Citation2, 32 Some examples of how replication fork progression and the replication program can be perturbed by exogenous or endogenous sources are described below.
Exogenous sources
Replication stress can be caused by agents that inhibit or block fork progression or cause DNA lesions. UV-light is an example of a physical agent that causes replication stalling by inducing pyrimidine dimers and blocking nucleotide incorporation by the replicative DNA polymerases.Citation43, 44 Aphidicolin is a natural inhibitor of B-family DNA polymerases, and may slow-down or arrest DNA replication, leading to reversible replication stress.Citation45-48 Chemicals used in anticancer chemotherapy and other genotoxic agents can cause DNA inter or intra-strand crosslinks, single-strand DNA breaks, base damage and DNA-protein crosslinks, that impede the elongation step of DNA replication.Citation2 Hydroxyurea (HU) is an anticancer agent that acts by inhibiting ribonucleotide reductase, the enzyme involved in nucleotide biosynthesis. HU is an example of how nucleotide depletion may lead to replication slow-down, change in origin usage, and progressive fork inactivation.Citation49, 50 Origin overactivation may also be caused by histone deacetylase inhibitors that affect the 3-dimensional conformation of chromatin.Citation51, 52 Other examples of chemicals causing replication stress are cisplatin and mitomycin C that induce DNA crosslinks, and camptothecin (or its derivatives topotecan and irinotecan) that inhibits Topoisomerase I activity.Citation2, 53
Endogenous sources
The cell is continuously facing a series of conditions that may limit its ability to replicate the entire DNA genome. Below is a list of several endogenous factors that may affect DNA replication and lead to genomic instability.
Difficult-to-replicate loci
These are genomic regions rich in di or tri-nucleotide repeats, palindromic sequences that tend to adopt cruciform structures, and G-rich sequences that form structures called G-quadruplexes (G4).Citation54-56 All these complex DNA structures constitute physical roadblocks to replication and can therefore act as replication fork barriers.Citation54,56,57 A good example of difficult-to-replicate loci is the telomeres, the ends of linear chromosomes, composed of tandemly repeated sequences organized in a specialized nucleoprotein structure that protects them from degradation and from being recognized as DSBs by the DNA repair machinery, and ensures their maintenance in face of the “end replication problem,” linked to the inability to fully replicate both strands of a linear DNA molecule.Citation58 Telomeric DNA may form G4 structures that are physiologically resolved by the DNA helicase RTEL1 and replicated with the help of Telomeric Repeat Binding Factor 1 (TRF1) and the homologous recombination proteins BRCA1 and 2.Citation59-61 Moreover, telomeres are replicated unidirectionally and are therefore more prone to replication pausing. Similarly, the ribosomal DNA (rDNA) represents an example of a highly unstable locus, due to its complex genomic organization, high transcription density, and unidirectional replication.Citation62-64 Studies on budding yeast rDNA have identified the existence of natural Replication Fork Barriers (RFBs), replication pause sites defined by specific DNA sequences that bind to non-nucleosomal proteins, blocking replication fork progression in the opposite direction to transcription and acting as recombinational hotspots.Citation65-67 RFB-mediated mechanisms at rDNA loci to block replication in case of converging transcription, preventing head-on collisions between replication and transcription, have been identified in many organisms, including murine and human cells.Citation64,68,69 Centromeres are also loci that are difficult to replicate due to their heterochromatic nature and richness in repetitive sequences.Citation70 Indeed, centromeric DNA forms looped structures and catenanes that are physiologically resolved by topoisomerase II α (TOP2A) in cooperation with the Bloom (BLM) protein complex.Citation71, 72 Finally, replication can be blocked by DNA-bound proteins, such as pre-Replication Complexes (pre-RCs) bound on dormant origins, which under normal conditions are removed by specialized helicases, like Rmr3 in budding yeast.Citation54, 73
Activated oncogenes
Different studies based on precancerous lesions have shown that replication stress, fork collapse and consequent genomic instability can be induced by the activation of oncogenes.Citation74-78 Replication stress and DNA damage following oncogene activation lead to a constitutive DNA damage response that induces cell senescence or apoptosis, thus protecting precancerous cells from becoming cancerous.Citation74, 79-81 There are different ways by which oncogene activation can lead to replication stress.Citation82 Activated oncogenes may alter the temporal program of replication and origin usage. For example, cyclin E activation leads to inhibition of pre-replication complex assembly and a decrease in origin firing, causing replication stalling and stress.Citation83 Contradictory with this finding, another study has shown that cyclin E overexpression impairs replication fork progression by inducing an excess of replication initiation and increasing conflicts between replication and transcription.Citation84 Deregulated origin firing following overexpression of several oncogenes may also lead to exhaustion of replication factors. Consistently, it has been shown that oncogene activation results in nucleotide depletion that in turn causes replication stress.Citation85 Following this work, Xie et al. have shown that Bcl-2 activation leads to ribonucleotide reductase inhibition and consequent replication stress.Citation86 Collectively, these studies suggest that oncogene-induced replication stress and DNA damage promote genomic instability and increase the selective pressure for mutations in p53 and DDR factors, which then allows cells to escape apoptosis and senescence, driving carcinogenesis.Citation75
Byproducts of cellular metabolism
Replication stress can also be caused as a result of DNA attack by byproducts of cellular metabolism.Citation87 The most important example of such products are free radicals including Reactive Oxygen Species (ROS) that may interfere with DNA replication. ROS can be the products of oncogene activation and can provide an alternative explanation for oncogene-induced replication stress.Citation76, 88 Oxidative stress-induced DNA damage or alteration of the nucleotide pool can induce fork slowing and replication stress, leading to genomic instability.Citation89 Another source of endogenous replication stress are reactive aldehydes, which are the byproducts of alcohol metabolism and histone demethylation and may cause DNA inter-strand crosslinks or protein-DNA crosslinks, blocking DNA replication.Citation90, 91 DNA lesions generated by reactive aldehydes have been shown to be critically dependent on a functional Fanconi anemia (FA) pathway for their repair, and deficiency in the enzymes that detoxify aldehydes act synergistically with FA pathway deficiency to induce genotoxic effects.Citation92
DNA conformation and chromatin
Unwinding of DNA by topoisomerases is a pre-requisite for initiation of DNA replication. Problems in DNA unwinding may therefore block the DNA polymerase complex and cause replication stalling.Citation93 It has also been shown that the elongation step of DNA replication occurs in parallel with the redeposition of histones and the reestablishment of marks on the newly synthesized DNA.Citation94 Therefore, defects in the process of chromatin assembly may block completion of DNA replication, leading to fork collapse.Citation95 A recent study has shown that there is a tight regulation between histone synthesis and DNA replication, mediated by the elongation factor PCNA (Proliferating Cell Nuclear Antigen).Citation96 Finally, chromatin compaction and structural features of heterochromatin may also constitute an impediment to DNA replication.Citation97, 98
Transcription-replication conflicts
Another major source of replication stress and genomic instability are encounters between the replication and transcription machineries.Citation62 The first method that eukaryotic cells adopt to avoid these collisions is spatial and temporal separation of the replication and transcription processes inside the nucleus.Citation99 If this separation does not occur, DNA polymerase progression can be blocked by the transcription machinery, leading to replication fork collapse and transcription-associated recombination.Citation100 A recent study has shown that the yeast ortholog of ATR, Mec1, together with the chromatin remodeler INO80 trigger the removal of RNA polymerase II from chromatin in HU-treated cells to avoid collisions between the transcription and replication machineries.Citation101 An independent study has also highlighted the role of INO80 in RNA polymerase II release from chromatin under conditions of transcriptional stress.Citation102 Interference between transcription and replication may also result in complementary binding between the newly synthesized RNA and one of the 2 replicating DNA strands.Citation103, 104 These RNA-DNA hybrids, called R-loops, are favored at CG-rich loci and are resolved by endogenous RNase H or specialized helicases, such as Senataxin and AquariusCitation105, 106. If not resolved, R-loops may lead to fork collapse and replication stress. RNA-DNA hybrids can form co-transcriptionally and are increased when transcription elongation or mRNA processing is impaired.Citation107-109 R-loops can also result from hybridization in trans of RNA molecules with complementary DNA, which in yeast is promoted by homologous recombination proteins.Citation110 R-loops have important functions in epigenetic regulation and gene expressionCitation111 but can induce genomic instability when they are deregulated or in excess.Citation112 Moreover, transcription may impede replication by inducing topological stress, which requires topoisomerase activity to prevent R-loop accumulation and genomic instability.Citation113, 114 Topological constraints can also be promoted by nuclear organization and higher order chromatin organization. For example, increased torsional stress accumulates at regions localized at the nuclear pores, where mRNA processing occurs.Citation115
Fragile sites
Chromosome fragile sites (FSs) are genomic regions that recurrently appear as gaps, breaks or constrictions on metaphase chromosomes.Citation116 The cytogenetic manifestation (or “expression”) of FSs was found to be induced in particular culture conditions, such as in folate- or thymidine-poor media.Citation117 FSs are classified as common or rare based on their frequency in the population.Citation116 Rare fragile sites are expressed in a small percentage of individuals (less than 5%), are inherited in a Mendelian manner, and their instability is associated with the expansion of trinucleotide repeats or AT-rich minisatellites; conversely, common fragile sites (CFSs) are present in all individuals, and thus, represent normal components of the chromosome structure that become unstable in replicative stress conditions. CFSs can be induced in vitro by exposing cells to low doses of aphidicolin or by altering the pool of nucleotides.Citation45, 118 They are located at late-replicating histone-hypoacetylated regions of the genome, and most of them are found within long genes, spanning from several hundred Kb up to 4 Mb in length.Citation116, 119-121 The sensitivity of these sites to breakage following replication stress is strongly dependent on cell type, suggesting that the DNA sequence is not sufficient to elicit their fragility, but the replication timing, the origin density, the transcription program and epigenetic factors may also define the characteristics of CFSs.Citation122-125
Several non-mutually exclusive mechanisms can explain the basis of CFS fragility at the molecular level. The first is based on the fact that these regions are located at late replicating domains comprising AT-rich sequences that can form secondary structures and may block or delay DNA replication.Citation116 Moreover, CFSs are characterized by a paucity of active replication origins so that, in conditions of replication slow-down, the neighboring forks have to travel long distances and may not be able to complete replication before mitosis.Citation28, 29, 122, 126 In addition, CFSs are often located within long genes that are transcribed over more than one cell cycle and their breakage is correlated with their transcription, indicating that CFS fragility can be induced by collisions between replication and transcription machineries and R-loop accumulation.Citation127 Indeed, Copy Number Variants (CNVs) that are often the outcome of chromosomal instability have been shown to overlap with CFSs and, in particular, with those localized at long, actively transcribed genes.Citation128 This would suggest that CNVs and CFSs are actually different manifestations of transcription-replication conflicts. Interestingly, it has been shown that transcription may displace MCMs bound to origins, causing a reduced number of licensed origins or inactivation of dormant origins, thus leading to origin paucity or to inefficient back-up of CFS replication in conditions of replication stress.Citation129-131 CFS instability is also induced by aberrant oncogene expression.Citation77, 132 The Kerem group has shown that different oncogenes elicit different fragility landscapes in the same cells, revealing the complexity of the mechanism that regulates the stability of CFSs.Citation125, 133 Interestingly, a recent study has identified a new class of fragile sites, called early-replicating fragile sites (ERFS), located in highly transcribed GC-rich repeat-containing genomic regions, causing many recurrent genome amplifications and deletions in B-lymphocytes,Citation134 further supporting a role for the interplay between replication and transcription in generation of fragility. Therefore, it would be of interest to understand whether breakage could be the result of persistence and/or mitotic processing of stalled forks or other types of intermediates formed as a consequence of replication-transcription conflicts.
Consequences of replication stress in mitosis
Despite the fact that replication fork impediments are normally sensed and managed by cells, endogenous or low levels of replication stress, induced, for example, by low doses of aphidicolin, may not be detected by the cell or may be tolerated, preventing the full activation of a checkpoint response.Citation3,135,136 In such cases, cells do not arrest in S-phase to complete replication but proceed into mitosis with under-replicated DNA or unresolved DNA structures.Citation12,13,15 Inactivation or dysfunction of dormant origins of replication induces a certain level of replication stress that may also lead to progression through mitosis with incomplete DNA replication.Citation27 These conditions result in persistence in mitosis of joint molecules (JMs) comprising under-replicated DNA, catenated DNA duplexes or unresolved DNA replication/repair intermediates that impede sister-chromatid disjunction.Citation24, 137 If not timely and safely processed, these latent JMs generate chromosome entanglements and segregation defects, leading to genomic instability. The most common consequences of replication stress that persist in mitosis are described below.
Multipolar mitoses
In addition to challenging genome duplication and chromosome disentanglement, endogenous or low levels of replication stress may also globally affect mitosis and chromosome segregation by inducing over-duplication of the centrosomes, leading to the assembly of mitotic spindles with multiple poles.Citation138-141 Interestingly, it was shown that supernumerary centrosomes arise spontaneously in homologous-recombination (HR)-deficient cells as a result of decreased fork speed and can be rescued by nucleoside supplementation.Citation140 Although the molecular link between replication stress and centrosome duplication remains unknown, an interesting study showed that Chk1 is localized at the centrosomes of interphase cells to prevent centrosome separation, activation of Cdk1 and entry into mitosis.Citation142 A prolonged S-phase that does not activate a full Chk1-dependent cell cycle arrest may, therefore, lead to centrosome over-duplication, with detrimental consequences in the following mitotic phase.
Anaphase bridges
Anaphase bridges are defined as DNA connections linking the segregating chromosomes during anaphase. Chromatin bridges or bulky anaphase bridges can be detected with classical DNA ligands, are histone-bound and represent the physical link between 2 incompletely separated sister chromatids or joined chromatids/chromosomes. Recently, a new class of anaphase bridges has been discovered, called Ultra Fine Bridges (UFBs).Citation143 UFBs are thin DNA bridges that, in contrast to the bulky bridges, cannot be stained with conventional DNA dyes (such as Hoechst or DAPI) and are devoid of histones. This might indicate that they contain a partially denatured, single-stranded, or stretched DNA conformation that is either not detectable or not permissive to intercalating agents.Citation144 UFBs were first observed by immunofluorescence staining of proteins coating these DNA threads in anaphase, specifically, the Bloom syndrome helicase (BLM) and the Plk1-interacting checkpoint helicase (PICH), which were shown to colocalize on those structures.Citation16, 19 A portion of these bridges is also bound by the ssDNA binding protein RPA.Citation13,145
There are at least 3 major classes of UFBs.Citation146 First, the centromeric UFBs (c-UFBs) that are usually formed after incomplete chromatid disjunction at centromeric regions and are marked by kinetochore proteins, such as Hec1.Citation12,16,19 The existence of these UFBs can be attributed to the complexity of the DNA sequences at these regions (see the section above) or to the increased centromeric chromatid cohesion, creating DNA catenanes that are difficult to resolve.Citation147 The c-UFBs are the most prevalent class of UFBs and are commonly observed in anaphase cells even in unperturbed conditions. They are induced by treatment with Topoisomerase II inhibitors, indicating that they most likely arise from incomplete decatenation of duplex DNA molecules.Citation19,148,149 Another class includes UFBs that do not have centromeric origin, but instead, they represent areas of DNA that have not been completely replicated or late replication/recombination intermediates that have not been resolved and preferentially arise at fragile site loci (fs-UFBs).Citation12,13 These UFBs are rarely observed in unperturbed conditions but are induced after treatment with agents that inhibit replication, such as aphidicolin or mitomycin C, and are characterized by the binding of the Fanconi anemia (FA) proteins FANCD2 and FANCI, not across the bridge but at their termini.Citation12, 13 Consistent with the role of replication stress and DNA damage in inducing this type of bridges, the tips of the fs-UFBs and their precursors are often positively stained for gammaH2AX.Citation12, 13 The third class of UFBs comprises the t-UFBs that derive from chromosome entanglements at telomeric loci. t-UFBs are rarely observed in normal conditions, but they are induced when proteins involved in telomere replication and maintenance are dysfunctional, such as the Werner syndrome helicase (WRN), the telomeric proteins TRF1 and TRF2, or the Taz1 fission yeast ortholog of human TRF1/TRF2.Citation150-153 Although the exact nature of the structures underlying UFBs is still unclear, it is thought that c-UFBs represent the physiologic manifestation of late decatenation of centromeric DNA and may contribute to generation of the proper tension between sister chromatids to promote correct segregation of chromosomes during anaphase,Citation149 while the other UFB forms are likely the result of under-replicated DNA that persists at difficult to replicate loci as a consequence of delayed replication or fork stalling in the presence of repetitive DNA, G-quadruplexes, R-loops, or tightly bound protein-DNA complexes, similar to what is reported for replication fork barriers in yeast.Citation145,154 Indeed, telomeres behave as fragile sites in the absence of TRF1, and telomere fragility can be induced by aphidicolin.Citation60, 155
UFBs may, therefore, derive from catenanes, hemicatenanes or other types of replication/recombination intermediates resembling Holliday junctions, which form independently from and are mostly prevented by RAD51.Citation13, 145 UFBs are bound at the transition between metaphase and anaphase by the BTRR complex, formed by the BLM helicase, Topoisomerase IIIa, RMI1 and RMI2, which likely act to resolve UFBs through unwinding of late replication intermediates, decatenation of hemicatenanes, or dissolution of recombination intermediates.Citation137
PICH is a DNA translocase with ATPase activity that is recruited to DNA after nuclear envelope breakdown in prometaphase.Citation19,144 PICH is needed during prometaphase and metaphase for proper chromosome condensation and architecture, and this function is distinct from its function during anaphase.Citation156,157 Interestingly, it has been shown that PICH binding to DNA is enhanced by tension-induced DNA stretching, which may explain the ability of PICH to bind to UFBs.Citation144 In addition, it has been shown that the ATPase activity of PICH is specifically required to prevent chromatin bridges but is dispensable for UFB resolution, suggesting that these 2 types of bridges may have different origins.Citation157 PICH has been proposed to function by preventing histone binding to areas that need to be repaired during mitosis, leading to the formation of UFBs.Citation156 A recent study has revealed that PICH has a role in attracting and activating the decatenase activity of TOP2A at highly repetitive loci, such as at rDNA and centromeric regions.Citation158 Following this work, it has been shown that, similar to centromeric DNA, rDNA may also undergo delayed decatenation and form PICH-stained anaphase bridges, which are induced by Topoisomerase II inhibition, thus identifying another class of UFBs.Citation159 Other clues regarding the nature and origin of UFBs have come from the identification of additional proteins that bind to these structures and are important for their resolution, including TOP2A, Topoisomerase IIβ-binding protein 1 (TopBP1), FANCM and Rif1. TopBP1, a protein involved in replication initiation, checkpoint activation and DNA repair, has been shown to bind and resolve anaphase bridges through an interaction with TOP2A.Citation145,160 The Fanconi anemia protein FANCM, a DNA translocase with branch migration activity, has also been shown to bind to UFBs, but only in late anaphase and telophase cells, where it replaces BLM, suggesting that it might have a role in resolution of persistent UFBs.Citation161 Interestingly, a recent study has shown that Rif1, a protein with multiple roles in replication timing, telomere replication and DSB repair,Citation162 colocalizes with PICH and BLM primarily on c-UFBs, but it has also been found to be recruited to APH-induced FANCD2-positive UFBs, suggesting that it localizes to UFBs independently from their origin.Citation163 In yeast, however, Rif1 has been shown to have a double-edged role in UFB processing because it prevents the resolution of t-UFBs while promoting the resolution of other non-telomeric UFBs, suggesting that UFBs of different origins may be channeled into different pathways or require different mechanisms for their resolution.Citation153
Under normal conditions, the majority of UFBs are resolved by late anaphase and telophase. Persistent UFBs may carry out some form of unresolved DNA damage or undergo breakage (either by mechanical stress at the cleavage furrow or by controlled endonuclease activity)Citation164 and transition into 53BP1 (p53 binding protein 1)-positive nuclear structures, called 53BP1 bodies, that are thought to shield DNA lesions in G1.Citation17,18,23 However, when not resolved in a timely manner or when they are in excess, persistent chromosome entanglements, chromatin bridges and UFBs can have detrimental consequences in terms of cell homeostasis and genome stability.
Rescue of under-replicated/unresolved DNA during mitosis
It is intriguing that, under endogenous or mild replication stress conditions, cells escape from checkpoint activation and proceed to G2, or even mitosis, with areas of incomplete replication.
It would be meaningful to determine why low levels of DNA damage or under-replicated DNA do not elicit cell cycle arrest,Citation3 whether threshold levels are tolerated, or whether a mechanism of adaptation is at work to allow cells to progress into mitosis, as initially proposed in yeast.Citation165-167 Interestingly, a recent work has shown that one essential function of ATR in unperturbed growth conditions is controlling the timing of cell cycle progression and preventing premature entry into mitosis, thereby keeping low threshold levels of under-replicated DNA at the onset of mitosis.Citation168 Indeed, it has been shown that yeast cells can proceed to anaphase with incomplete DNA replication, even in the presence of an active checkpoint.Citation169 Likewise, a series of recent studies in metazoans have shown that some under-replicated chromosomal regions remain, even after entering into mitosis. The cells have developed different mechanisms to resolve these structures during mitosis to prevent chromosome mis-segregation.Citation15 Below, we describe the mechanisms and pathways that have recently been determined to play a role in the resolution of under-replicated DNA during mitosis.
FA pathway and its roles in the replication stress response
FA is a rare genetic disease characterized by developmental abnormalities, bone marrow failure and cancer predisposition, caused by mutations in at least 20 FANC genes.Citation170-172 The FA proteins constitute a pathway involved in the replication stress response and repair of interstrand crosslinks (ICL). Most FANC proteins form the FA core complex that primarily acts as a ubiquitin ligase to mono-ubiquitinate FANCI and FANCD2, which form a heterodimer called the ID complex and promote ICL repair by the coordinated action of endonucleases, TLS polymerases and homologous recombination proteins. The cells of FA patients display hypersensitivity to ICL and an increased frequency of spontaneous and ICL-induced chromosomal aberrations and radial chromosomes.
Recent works have shown that FA proteins, apart from their role in ICL repair, have an important role in maintaining genomic stability after replication stress, or even during normal replication, by regulating origin firing, replication fork stability and restart. FANCD2 and HR proteins have been shown to stabilize replication forks after HU treatment and protect nascent DNA from nucleolytic degradation.Citation173 In addition, FANCD2 and FANCI have been isolated on nascent DNA at stalled replication forks,Citation174,175 and FANCI also binds to unperturbed, active forks.Citation175 FANCD2 interacts with MCM proteins, independently from its monoubiquitination by the FA core, and restrains DNA synthesis in conditions of nucleotide deprivation, thus preventing ssDNA accumulation and promoting recovery from replication stress.Citation174 Moreover, it has been shown that FANCI and FANCD2 have independent opposing roles in conditions of low replication stress: FANCI is required for activation of dormant origins to enable the timely completion of DNA replication before mitosis, whereas FANCD2 inhibits origin firing and restrains DNA synthesis.Citation176 The functions of these proteins are ATR-dependent because, in conditions of high replication stress, ATR phosphorylates FANCI to allow binding to FANCD2 and suppression of origin firing. The FANCD2-FANCI heterodimer then binds to chromatin and is monoubiquitinated to promote fork repair and restart.Citation176-178 In addition, recent studies have highlighted the role of HR and FA proteins in regulating the conflicts between replication and transcription.Citation179-183 FA-deficient cells were shown to accumulate R-loops and DNA breaks, which could be prevented by RNase H or transcription inhibitors.Citation182,183 Garcia Rubio et al. also showed that FANCM translocase activity was required for R-loop suppression.
Consistent with its role in preventing or resolving replication problems, the FA pathway regulates CFS replication and stability.Citation184,185 The function of FA proteins extends beyond S-phase.Citation15 If recovery from replication stress in S-phase is not successful or incomplete, FANCD2 and FANCI persist at CFSs on mitotic chromosomes even up to telophase.Citation12,13 Although the function of FA proteins in mitosis remains unknown, they have been shown to promote BLM-mediated resolution of aphidicolin-induced, non-centromeric (Hec1-negative) anaphase bridges, limiting chromosome mis-segregation and aneuploidy.Citation12 In addition, FANCM is recruited to UFBs in late telophase and likely contributes to bridge resolution.Citation161 Although it is not clear how FA proteins fulfill their role, the functional crosstalk between the FA pathway and BLM in response to replication stress may be important in both S-phase and mitosis.Citation12,186-188 During mitosis, BLM exerts its function in concert with Topoisomerase IIIα to promote complete sister chromatid disjunction and UFB resolution.Citation16 Because FANCD2 and FANCI do not bind across bridges but to their extremities, they might have a role in stabilizing the DNA structure or regulating the chromatin environment in a way that promotes UFB resolution. Further work will be required to solve this issue.
Structure-selective endonucleases and their role at fragile sites
The XPF/MUS81 family of structure-selective endonucleases are nucleolytic enzymes that recognize and cleave specific DNA structures instead of DNA sequence elements.Citation189 MUS81 is the catalytic subunit of a cell cycle-regulated structure-selective endonuclease involved in interstrand crosslink repair, homologous recombination and replication fork restart. It forms a complex with EME1, its regulatory subunit, and the MUS81-EME1 complex acts by cutting 3′ flaps, D-loops, branched DNA structures and Holliday junctions. ERCC1, which forms a complex with XPF, is another structure-selective endonuclease that is involved in nucleotide excision repair and interstrand crosslink repair, cleaving bubble-like structures, stem-loops and 3′ flaps. In mouse embryonic stem (ES) cells, Mus81-Eme1 has been shown to bind to stalled replication forks and induce DNA breaks that are essential for the recovery of the forks.Citation190
Recently, 2 studies have revealed an interesting role for MUS81-EME1,Citation20 21 and ERCC1Citation20 in processing under-replicated or unresolved DNA structures at CFSs in mitosis. Specifically, these studies have shown that, following mild replication stress, these endonucleases are recruited and cut DNA at CFSs to resolve replication intermediates and prevent the formation of anaphase bridges. They are recruited to CFSs and colocalize with FANCD2 foci from late G2 or early prophase, when the chromosomes start to condense, until the metaphase to anaphase transition,Citation20,21 suggesting that their action is required to release chromosome interlinkage before anaphase. Intriguingly, depletion of MUS81 and ERCC1 was associated with a decreased frequency of CFS gaps and breaks in metaphase but a concurrent increase in chromatin bridges and UFBs, as well as mitotic catastrophes and accumulation of DNA breaks and 53BP1 bodies in G1, indicating that, even if breakage at CFSs in mitosis can potentially lead to genetic instability, tightly controlled endonuclease activity of MUS81 and ERCC1 prevents chromosome mis-segregation and transmission of DNA damage to daughter cells.Citation20, 21
In addition to the endonucleases described above, subsequent works have revealed a role for the scaffold protein SLX4 during mitosis in regulation of CFS stability and prevention of anaphase bridges, via its newly reported SUMO-binding,Citation191, 192 and SUMO ligase activity.Citation191 SLX4 is an FA protein that serves as a binding platform for MUS81-EME1 and XPF-ERCC1 and has an essential role in ICL repair.Citation193 Guervilly et al. showed that SLX4 colocalizes with MUS81 and ERCC1 in mitotic cells and is required for their recruitment, which suggests that SLX4 acts in the same pathway as the MUS81-EME1 and XPF-ERCC1 endonucleases and promotes controlled DNA processing at CFSs before anaphase, to prevent chromosome mis-segregation and mitotic catastrophe.Citation191
Unscheduled DNA synthesis in late G2 to early mitosis
Several studies have demonstrated that the cell is able to resolve incompletely replicated DNA structures that persist in mitosis and that this can occur even at the last-minute, just before entry into anaphase, to prevent chromosome mis-segregation and its detrimental consequences. By pulse-labeling cells with the thymidine analog ethynyldeoxyuridine (EdU), the Hoffmann and Rosselli groups have shown that active DNA synthesis can occur in late G2 or early mitosis.Citation6, 20 The EdU incorporation was observed, though very rarely, even in unperturbed mitotic cells but was significantly induced after treatment of cells with low-doses of aphidicolin.Citation6 Moreover, this type of unscheduled DNA synthesis was increased in cells deficient for the TLS polymerase Polη, which was shown to contribute to CFS replication in S-phase, preventing CFS instability.Citation6 The majority of sites showing EdU incorporation in mitotic cells corresponded to the chromosome loci targeted by FANCD2 and endonucleases, indicating that completion of DNA replication at CFSs and possibly other genomic regions can be delayed until mitosis.Citation6, 20 In line with these findings, several independent studies have shown that replicative or repair DNA synthesis at late replicating or challenging loci in early mitosis represents a last attempt to complete genome duplication and limit under-replicated DNA and mitotic defects.Citation22, 23, 168, 194-196
TopBP1-dependent mitotic DNA synthesis
The Lisby laboratory has demonstrated the existence of active unscheduled DNA synthesis in mitosis, by showing Topoisomerase IIβ-binding protein 1 (TopBP1)-dependent incorporation of nucleotides in mitotic cells.Citation23 The authors showed that TopBP1 colocalizes with FANCD2 foci in early mitosis after mild replication stress and is required for SLX4 binding to chromatin. TopBP1-dependent DNA synthesis is known to occur after mitotic onset because a mutant TopBP1 protein lacking the nuclear localization signal, therefore only capable of accessing chromatin after breakdown of the nuclear envelope, rescued the reduced EdU incorporation observed in TopBP1-deficient cells.Citation23 Taken together, these data suggest that the cell is able to complete DNA synthesis in mitosis, in the presence of condensed chromatin, at regions of delayed replication forks. Interestingly, TopBP1 not only localizes with the APH-induced FANCD2-targeted sites but also forms spontaneous foci at other chromatin loci in mitotic cells independently from replicative stress; even if these structures do not seem to be linked to replication stress, TopBP1-mediated resolution is necessary to prevent 53BP1 nuclear body formation and transmission of DNA damage to daughter cells.Citation23
Break-induced replication (BIR)
Recently, the Hickson group has shown that replication stress induces POLD3-dependent DNA synthesis in mitosis, which is promoted upon cleavage of replication forks at CFSs by the MUS81-EME1 in complex with SLX4.Citation22 Specifically, by showing EdU incorporation at CFSs in mitotic cells after a G2-block, they demonstrated a chromatin condensation-dependent mechanism of DNA synthesis that occurs during the transition of cells from G2 to prophase and early prometaphase. POLD3 (pol32 in yeast) is a regulatory subunit of DNA polymerase δ required for break-induced replication (BIR), a well conserved HR-mediated mechanism that utilizes a single-ended DSB to restart replication at broken or collapsed forks and to maintain eroded telomeres.Citation197-199 BIR has been most well characterized in yeast, where it has been shown to comprise different sub-pathways that can be Rad51-dependent or independent.Citation197,198 In contrast to the canonical HR pathway, BIR is an error-prone type of HR, often based on very short sequences of homology and prone to fork stalling and template switching events (called micro-homology mediated BIR or MMBIR or FoSTeS), leading to chromosome duplications, copy number variations (CNVs) and gross chromosomal rearrangements (GCRs).Citation197,200 How this pathway is regulated during mitosis is not known. It has been shown that CFS-associated intermediates form independently of RAD51,Citation13,139,201 and that HR proteins, such as BRCA1, RAD51 and BLM, are inactivated by CDK1-mediated phosphorylation and dissociate from the chromatin by late G2.Citation12,13,202 In addition, DSB repair in mitosis has been shown to be inhibited to avoid aberrant joining of chromosome ends.Citation203,204 This suggests that BIR-mediated rescue of stalled forks at CFSs in mitosis would rely on a RAD51-independent pathway, likely as a backup mechanism.Citation205
The activity of MUS81 is increased through phosphorylation of its binding partner EME1 by CDK1 in G2-M, providing a way to fine tune regulation of its enzymatic activity.Citation206 Indeed, it has been shown that oncogenic stress induces premature MUS81 activation, leading to aberrant processing of reversed replication forks and generation of toxic replication intermediates.Citation207 Endonucleases may participate in multiple steps during BIR to generate or displace the strand invasion intermediate, cleave flap structures, restore an active fork and resolve recombination intermediates.Citation208 Work in yeast has shown that the BIR pathway can be promoted in the absence of Mus81 and Yen1 endonucleasesCitation209 and that Mus81 may restrain BIR and suppress template switching.Citation210 Therefore, it would be interesting to determine how the endonuclease complexes are recruited and regulated and whether other pathways participate in rescue from replication stress at CFSs and possibly other loci during mitosis. In this context, SLX4 and its SUMO-related functions may be a central regulator of MRRS.Citation211 During ICL repair, the FA pathway has been shown to promote endonuclease recruitment to damaged DNA and ICL unhooking.Citation212 However, MUS81 and ERCC1 recruitment to mitotic chromosomes has been shown to occur independently of FA pathway activation.Citation20 Using chicken DT40 cells deficient for FANCD2, Pedersen et al. also showed that SLX4 foci assembly in mitosis is independent from FANCD2.Citation23 Interestingly, TopBP1 was shown to be required for SLX4 recruitment and mitotic DNA synthesis.Citation23 However, it is still unclear whether DNA synthesis at these sites occurs by a homologous recombination (HR)-mediated mechanism or by simple loading of DNA polymerases. Since chicken cells lack a MUS81 ortholog,Citation189 another endonuclease or another mechanism must be responsible for mitotic synthesis in these cells.
Translesion synthesis (TLS)
TLS is a damage tolerance mechanism alternative to HR that addresses replication stress induced by stalling of the replicative DNA polymerases at various lesions on template DNA.Citation213-215 It is an error-prone process that allows DNA replication by directly bypassing the DNA lesions. It is mediated by specialized low-fidelity DNA polymerases, such as Polη and Polζ, that are recruited to chromatin by monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) and the polymerase scaffold protein Rev1.Citation216,217 A study by the Hoffmann laboratory showed that Polη is required for DNA synthesis at CFSs during S-phase, to allow timely completion of DNA replication before entry into mitosis.Citation6 In line with this work, a study by the Kupfer group showed that a non-ubiquitinated form of FANCD2, in a complex with RAD51 and the E3 ubiquitin ligase Rad18, is required for PCNA monoubiquitination and Polη assembly onto chromatin after HU-induced replication stress,Citation218 which may suggest possible crosstalk between FANCD2 and Polη in facilitating CFS replication. Moreover, a previous study demonstrated the role of Rev3, the catalytic subunit of Polζ, in preventing CFS instability, suggesting that Rev3-mediated DNA synthesis is activated during G2-M phase to allow completion of replication at CFSs before chromosome segregation.Citation194 On the other hand, a recent study by Gallina et al. demonstrated that mitotic DNA synthesis is independent of DNA Polη and TLS.Citation219 Specifically, this study showed that unscheduled DNA synthesis in mitosis is independent of Rev1 and ubiquitinated PCNA in DT40 chicken cells and that Polη does not colocalize with either TopBP1 or FANCD2 foci in mitotic cells, indicating that nucleotide incorporation in mitosis is not due to the TLS pathway. Taken together, these studies suggest a putative but unclear role for TLS polymerases in the mechanism that leads to resolution of incomplete DNA replication that persists in late G2 to mitosis. Recent studies suggest that specialized polymerases, apart from their role in bypassing damaged DNA templates, may participate in replication across non canonical DNA structures and difficult to replicate loci.Citation220 Interestingly, in yeast, Pol31 (POLD2) and Pol32 (POLD3) accessory subunits of Pol δ have been shown to copurify with the Rev3 and Rev7 subunits of Polζ to form a 4-subunit Polζ complex, leading to the hypothesis that the Polζ holoenzyme may replace Pol δ at replication blocking lesions or perform TLS or other PCNA-dependent cellular functions.Citation221-223 Moreover, the TLS polymerases polζ and Rev1 have been recently shown to promote template switching during MMBIR, leading to genome rearrangements.Citation224 Therefore, it is tempting to speculate that specialized polymerases may promote replication or replication-associated repair pathways at challenging loci, which can both prevent or promote genome instability.
It has been proposed that the appearance of gaps and breaks on mitotic chromosomes (CFS expression) is the cytogenetic manifestation of mitotic repair synthesis taking place at these sites, to limit chromosome non-disjunction and mis-segregation.Citation22 However, inhibition of mitotic DNA synthesis may not always be associated with a decreased CFS expression.Citation23, 194 Indeed, TopBP1 deficiency impairs mitotic synthesis but is associated with an increased CFS expression.Citation23 Moreover, mitotic synthesis and CFS expression are not systematically associated with the rescue of anaphase bridges. For instance, ATR- or FA-deficient cells display both an increased CFS expression and mitotic synthesis, together with an increased frequency of anaphase bridges.Citation12,161,168,184,195,225 In BLM-deficient cells, the downregulated expression of cytidine deaminase (CDA, an enzyme involved in the pyrimidine salvage pathway) leads to an increase in the dCTP pool and reduced PARP-1 activity, with a consequent increase in mitotic DNA synthesis and UFB frequency.Citation196 Therefore, delayed DNA synthesis may not be sufficient to complete replication, leaving specific loci incompletely replicated. The pool of nucleotides is regulated during the cell cycle; therefore, unscheduled DNA synthesis may be inefficient or inaccurate.Citation226 Alternatively, rescue of under-replicated DNA during mitosis may result in an excess of replication or repair intermediates that overwhelm the resolution pathways. Therefore, we favor the view that CFSs represent replication termination zones where processing of replication or repair intermediates is still ongoing or incomplete, preventing the proper resolution of topological constraints and chromatin organization.
Consequences of failed rescue of replicative stress in mitosis
Absence of rescue of replicative DNA damage in mitosis can have detrimental consequences for cells, as it may lead to tumorigenesis. However, healthy cells are able to prevent carcinogenesis by activating the p53 pathway that leads to apoptotic cell death.Citation227 Alternatively, in primary cells, for example, replication stress can induce a response that leads to proliferation and cell growth arrest, a phenomenon called cellular senescence.Citation228 Cell death in mitosis and formation of 53BP1 nuclear bodies are the primary mechanisms that protect cells with persisting damage or under-replicated DNA in mitosis from becoming carcinogenic.Citation17, 229 However, despite the efficiency of these mechanisms, cells can be defective in one of these pathways and, therefore, become micronucleated or aneuploid, leading to tumorigenesis.
Mitotic catastrophe
Cell death in mitosis, also known as mitotic catastrophe, is the result of failure to complete mitosis.Citation229 It usually occurs as a consequence of serious problems of nondisjunction or after centrosome overduplication and consequent entry into mitosis with multiple spindle poles.Citation230 Although there are several studies linking apoptosis to mitotic catastrophe, it is still unclear whether mitotic catastrophe is an apoptotic mechanism.Citation231 However, mitotic catastrophe remains a cell death mechanism that can eliminate cells with persistent replicative DNA damage and prevent carcinogenesis.
53BP1 bodies
53BP1 is a DNA repair marker that colocalizes with γH2AX at DSBs. It has been recently shown that 53BP1 makes foci at persisting DNA lesions induced by replication stress.Citation17, 18 Specifically, 53BP1 forms large nuclear structures called “bodies” in the subsequent G1-phase of the cell cycle. These bodies preferentially associate with CFSs and are largely induced in the absence of BLM.Citation17 The biologic function of 53BP1 bodies could be to shield DNA lesions that remain unrepaired during the transition from G2 to mitosis, so that they are efficiently repaired in the next cell cycle.Citation17 Recently, it has been shown that 53BP1 bodies contain ssDNA marked by RPA and increase following MCM depletion, due to the presence of large replicons devoid of origins.Citation232 This finding is consistent with the fact that 53BP1 bodies occur due to under-replicated DNA persisting in mitosis and are enriched at CFSs following replication stress. 53BP1 bodies are also stained by DSB markers, which can result from breakage of under-replicated DNA or unresolved DNA structures at the end of mitosis.Citation20,21,233 53BP1 bodies may also be associated with other types of DNA or chromatin alterations that are transmitted in G1, which depend on TopBP1 for their resolution in mitosis.Citation23 There are 2 most likely functions for 53BP1 in the cell cycle following DNA damage. The first is promotion of Non-Homologous End Joining (NHEJ) that occurs in G1,Citation234 which could, however, be error-prone and promote inappropriate joining of DNA ends, leading to chromosomal rearrangements.Citation235 The second possibility is that it shields the damaged sites from excessive resection and aberrant repair in G1 and protects them until entry into S-phase, when they can be repaired by HR-mediated mechanisms.Citation17 The latter possibility is supported by the fact that 53BP1 bodies stay in the nucleus up until entry of the cell into the subsequent S-phase. 53BP1 may allow for repair of some lesions, while protecting complex DNA lesions from further damage or error-prone repair and, therefore, prevent carcinogenesis. Further work would be required to know whether these large nuclear bodies mark clustered DNA lesions or chromatin domains and to have a better understanding of the function of 53BP1 in the cell cycle following replication stress.
Non-disjunction and aneuploidy
Failure to recover from replication stress and to resolve joint molecules that persist in mitosis can also result in chromosome non-disjunction and mis-segregation, leading to gain or loss of chromosome fragments or whole chromosomes.Citation12 The latter situation, known as aneuploidy, is a common cancer trigger and may explain why patients with mutations in genes regulating replication stress are often susceptible to cancer.Citation14,236 Interestingly, it has been shown that supernumerary chromosomes can generate additional replication stress, thus precipitating genomic instability.Citation236 In addition, aneuploidy can induce changes in gene expression and proteotoxic stress, leading to deregulation of cellular functions and homeostasis.Citation237-239
Micronuclei
A frequent outcome of the absence of rescue of replicative stress in mitosis is the formation of small chromatin masses called micronuclei.Citation12 These small nuclear fragments, also known as Howell-Jolly bodies, represent lagging chromosomes or chromosome fragments that were unable to properly segregate with the rest of the genome but were efficiently nucleated after the end of mitosis.Citation240 Replication of these micronuclei in the subsequent S-phase is abnormal and may lead to further chromosomal aberrations. The micronuclei can eventually fuse with the main nucleus, leading to large chromosomal abnormalities, or become shattered into small pieces, a phenomenon called chromothripsis.Citation241 In both cases, the cells accumulate serious irreversible genome modifications, such as chromosome deletions, triplications, inversions and translocations, which may subsequently lead to cancer.
Cytokinesis failure
Another consequence of replication stress and failure to resolve chromosome entanglements is impairment of cytokinesis. Indeed, persistent anaphase bridges and UFBs have been shown to inhibit abscission and result in binucleated cells.Citation23,149,161 The presence of lagging chromosomes or chromatin bridges in telophase has been shown to activate a pathway called NoCut in yeast and an AuroraB-dependent abscission checkpoint in higher eukaryotes to delay cytokinesis and inhibit abscission, allowing the cell to segregate all the genetic material.Citation242-244 Defective activation of this pathway can lead to breakage of chromosomes trapped in the spindle midzone or to furrow regression and tetraploidy. Conversely, delay or failure to resolve anaphase bridges and persistent activation of this pathway may impair abscission. Anaphase bridges and lagging chromosomes following replication stress can also lead to chromosome breakage and aneuploidyCitation12 and may be the cause of numerical and structural chromosomal rearrangements in tumor cells.Citation14 A recent work has shown that chromatin bridges induced by replication stress are able to activate the NoCut pathway, leading to cytokinesis delay, while lagging chromosomes or anaphase bridges resulting from dicentric chromosomes do not activate this pathway, and thus, lead to aneuploidy and chromosome breakage.Citation245 This may explain the different fates of anaphase bridges and lagging chromosomes arising from different causes, and it suggests that structural and numerical chromosome aberrations after replication stress may be the result of dicentric chromosomes or chromosome structures that are severed during anaphase or cytokinesis and/or do not activate this checkpoint.Citation235,246-248 Indeed, ATR and chk1 have been shown to link replication stress with the AuroraB pathway and to regulate the timing of abscission at the midbody stage.Citation249 Interestingly, ATR-deficient cells undergoing mitosis with under-replicated DNA show segregation defects and cytokinesis failure, which can be mitigated by partial inhibition of Cdk1 activity.Citation168 Therefore, a tight coordination between under-replicated DNA processing and cytokinesis may be crucial for timely resolution of chromosome entanglements and abscission.
Epigenetic instability
Another outcome of replication stress and DNA damage is perturbation of histone recycling and restoration of chromatin structure, which can affect epigenetic maintenance and cell function.Citation97,250-254 For example, replication fork blockage at G-rich genomic sequences has been associated with a biased incorporation of H4-acetylated histones and loss of repressive chromatin marks.Citation255 Conversely, uncoupling of DNA replication with new histone deposition has also been shown to alter predeposition marks on new histones and induce heterochromatinization and gene silencing.Citation256 Likewise, trinucleotide repeat expansion was shown to mediate gene silencing.Citation257 Moreover, inhibition of DNA synthesis may also alter DNA methylation.Citation258 Persistence of under-replicated DNA in mitosis may therefore interfere with normal chromatin reassembly and propagation of the epigenetic information. Indeed, UFBs are largely devoid of histones and therefore, need chromatin structure and epigenetic information to be re-established in the next cell cycle. How the cells cope with epigenome maintenance after persistent replication stress in mitosis is largely unknown. Interestingly, recent studies have shown that Drosophila male germline stem cells that divide by asymmetric cell division differentially distribute old and new histones, and selectively segregate the parental histones into the stem cell and the newly synthesized histones into the differentiating daughter cell.Citation259 One could speculate that differential histone incorporation following replication stress or DNA damage may allow discrimination between daughter cells and determine cell fate.Citation252
The role of mitotic replication stress in human disease
The most common human disease associated with defective rescue of replication stress is cancer.Citation260 The inability to repair fragility at CFSs or anaphase bridges persisting into telophase leads to chromosome mis-segregation, micronuclei formation and aneuploidy, which are often present in cancer cells. Cells with micronuclei or an abnormal number of chromosomes become carcinogenic, either by losing the expression of tumor suppressors or by activating the expression of oncogenes. These transformed cells then lose their proliferation control and create tumors that eventually lead to organ failure.Citation2
A common characteristic of many cancers is the genomic instability observed at CFSs. CFS breakage may induce recombinogenic events, viral integrations, and chromosomal rearrangements.Citation116 Indeed, recent analyses of cancer genomes have revealed that CFSs are involved in the majority of recurrent chromosomal deletions and translocations in cancer.Citation261-263 Importantly, CFSs represent the preferential targets of oncogene-induced replication stress and are involved in the formation of DSBs that contribute to genomic instability in the earliest stages of tumor development.Citation77 However, the contribution of CFSs to the tumorigenic process is still under debate.Citation264 Molecular characterization of CFSs has shown that many of them map within or in proximity to cancer-associated genes, which are aberrantly expressed in several tumors.Citation265,266 One of the most well-studied CFSs, FRA3B, which shows a high frequency of breakage in lymphocytes and is frequently abnormal in epithelial tumors,Citation263,267 is located within the Fragile Histidine Triad (FHIT) gene. The protein encoded by FHIT is involved in the DNA damage response, nucleotide metabolism and apoptosis.Citation116,268 Loss of FHIT expression induces replication stress and accumulation of DNA damage, leading to genomic instability and promotion of tumorigenesis.Citation268 Other CFSs that overlap with known tumor-suppressor genes are FRA16D, located within a gene encoding for the multifunctional enzyme WW-domain containing oxidoreductase (WWOX), and FRA6E, located within the PARK2 gene encoding the E3 ubiquitin ligase that is mutated in Parkinson disease, which has been shown to regulate cyclin turnover.Citation269-272 Breakage at these sites, resulting in loss-of-function of these tumor-suppressor genes, provides a possible explanation of how CFS instability can contribute to cancer initiation and progression. In addition, CFS expression can promote the amplification of oncogenes located in their proximity via breakage-fusion-breakage mechanisms, as described, for example, for MET at FRA7G or PIP at FRA7I.Citation273-275
Apart from cancer, many known syndromes are associated with mutations in genes involved in the rescue of mitotic replication stress.Citation2 Seckel syndrome, for example, a developmental disease characterized by dwarfism, microcephaly and mental deficiencies, is induced by mutations in ATR or ATRIP. Fanconi anemia is another example of such a disease; individuals with mutations in FA genes are characterized by a wide range of symptoms from skeletal and developmental defects to cancer predisposition and progressive bone marrow failure.Citation276,277 Indeed, FA cells show a high frequency of abnormal chromosome structures that may occur as a result of persistent replication stress during mitosis. FA patients display chromosomal aberrations at CFS loci.Citation278-281 Importantly, persistent anaphase bridges have been observed in haematopoietic cells of FA mice, even in unperturbed conditions and are associated with cytokinesis failure and increased apoptotic cell death,Citation161 suggesting that endogenous replication stress or unresolved DNA damage that persists in mitosis may contribute to bone marrow failure and account for the exacerbated p53/p21 activation observed in FA.Citation171 Recently, the FA pathway has also been shown to counteract physiologic stress during megakaryopoiesis, preventing CFS instability and cell division abnormalities associated with defective megakaryocyte differentiation and thrombocytopenia of FA mice.Citation282 Finally, a variant form of xeroderma pigmentosum (XPV) is a cancer-prone disease that is caused by mutations in the gene coding for the TLS polymerase Polη, a protein shown to have a role in DNA synthesis at CFSs.Citation6
Deficiency in BLM protein leads to a disease that has similar symptoms to FA but is also characterized by immunodeficiency and premature aging.Citation283 Aging is prematurely induced in syndromes characterized by mutations in RecQ DNA helicases, such as the Werner (WRN) helicase (Werner's syndrome) or the RecQL4 helicase (Rothmund-Thomson syndrome)[283]. Apart from BLM, WRN has also been shown to have a functional role in preventing replication stress by regulating CFS stability.Citation284 These syndromes indicate that the inability to properly and timely resolve replication challenges at CFS, telomeres and/or other difficult-to-replicate loci may affect overall cellular morphology and homeostasis and eventually lead to aging.
Interestingly, replication stress may also be associated with neurodevelopmental and neurodegenerative disorders.Citation121 Epilepsy, schizophrenia and autism are neurologic syndromes associated with genomic CNVs, which might occur as a result of defective repair at CFSs. Indeed, a recent study has shown that long genes with neural function are hotspots of recurrent DSB clusters (RDCs) in neuronal stem cells.Citation285 Most of these RDCs were induced by low-dose APH treatment and mapped within long, transcribed and late-replicating genes associated to CFSs. It would be important to better understand the link between the expression of these genes and their fragility, and to determine whether somatic CNVs in these genes can modulate neuronal function and plasticity. Moreover, mutations in the gene encoding for Senataxin (SETX), a protein involved in resolving collisions between transcription and replication and, therefore, preventing R-loop formation, lead to various neurodegenerative diseases, such as amyotrophic lateral sclerosis 4 and ataxia-ocular apraxia 2.Citation106,286 The molecular link between neurologic diseases and replication stress may lie in the fact that many of the genes involved in brain development are long and are therefore more susceptible to collisions between transcription and replication,Citation127 transcription-associated DNA damage, or replication stress-induced segregation defects and aneuploidy.Citation287
Concluding remarks
Replication stress is one of the most important molecular triggers of carcinogenesis. Our knowledge of the DNA damage response to decelerated or aborted DNA replication has dramatically expanded during the last 2 decades. Although the replication stress response is largely constrained within S-phase, with the help of the ATR/Chk1-induced intra S-phase checkpoint, recent studies have shown that endogenous or mild replication stress can lead to the persistence of under-replicated or unresolved DNA structures in mitosis. This persistence can have detrimental consequences for the cell, as it may lead to chromosome nondisjunction and other aberrant mitotic events that can limit cell survival or alter normal cellular function and organismal development. In addition, it could also drive genetic and epigenetic instability, promoting cancer initiation and progression. We have just started to identify the mechanisms by which the cell is able to resolve this persistent replication stress in mitosis and some of the key factors and pathways involved (). Although the role of these proteins has been partially characterized, we still do not know how chromatin binding or the activity of these proteins is regulated, the intermediates they act on, or how these factors cooperate to resolve replication stress before the end of mitosis. We also expect that other factors that will contribute to our understanding of MRRS await identification and may provide important clues to the mechanisms involved in the development of disease, including cancer. A major challenge of future studies will be to understand how transcription and RNA metabolism regulators interact with replication and repair machineries to prevent or resolve potential conflicts between these metabolic processes and how they are coordinated with chromatin modifications and chromosome architecture during mitosis to ensure accurate transmission of the genetic and epigenetic information to daughter cells.
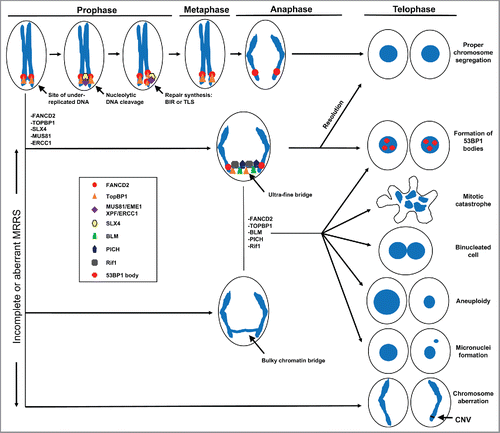
Figure 1. Schematic diagram summarizing the cellular response to under-replicated DNA that persists in mitosis. FANCD2 and TopBP1 bind at sites of incomplete DNA replication during late G2/early prophase. Then, with the help of SLX4, the endonucleases MUS81 and ERCC1 bind at these sites and mediate nucleolytic cleavage of replication intermediates. This allows repair synthesis to occur to complete DNA replication and/or resolution of joint molecule intermediates, promoting proper chromosome segregation. In the absence of FANCD2, TopBP1, SLX4, MUS81 or ERCC1, a bulky or ultra-fine anaphase bridge (UFB) may form from interlinked or under-replicated DNA. The UFB may be resolved before completion of anaphase with the help of FANCD2, TopBP1, PICH, Rif1 and BLM, or transition into 53BP1 bodies in the daughter cells. However, unresolved joint molecule intermediates, persistent UFBs and bulky chromatin bridges, lead to chromosome disjunction, segregation defects and cell division abnormalities, which result in mitotic catastrophe, binucleation, aneuploidy or formation of micronuclei. Finally, nucleolytic cleavage and aberrant repair of under-replicated DNA in prophase may also lead to chromosomal deletions, amplifications or rearrangements that have detrimental consequences for the cell.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We apologize for all the work and primary references we could not cite due to space limitations. We thank the members of the V. Naim team for helpful discussions on the subject of this review. The authors are very grateful to Michael Lisby, Vibe H. Oestergaard and Benoit Miotto for critical reading of the manuscript.
Funding
Research in the V. Naim laboratory is supported by a European Research Council Starting Grant (ERC-2014-StG-638898 “FAtoUnFRAGILITY”).
Related Research Data
References
- Sirbu BM, Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 2013; 5(8):a012724; PMID:23813586; http://dx.doi.org/10.1101/cshperspect.a012724
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16(1):2-9; PMID:24366029; http://dx.doi.org/10.1038/ncb2897
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300(5625):1542-8; PMID:12791985; http://dx.doi.org/10.1126/science.1083430
- Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst) 2009; 8(9):1038-46; PMID:19482564; http://dx.doi.org/10.1016/j.dnarep.2009.04.014
- Jossen R, Bermejo R. The DNA damage checkpoint response to replication stress: A Game of Forks. Front Genet 2013; 4, 26; PMID:23493417; http://dx.doi.org/10.3389/fgene.2013.00026
- Bergoglio V, Boyer AS, Walsh E, Naim V, Legube G, Lee MY, Rey L, Rosselli F, Cazaux C, Eckert KA, et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J Cell Biol 2013; 201(3):395-408; PMID:23609533; http://dx.doi.org/10.1083/jcb.201207066
- Le Beau MM, Rassool FV, Neilly ME, Espinosa R 3rd, Glover TW, Smith DI, McKeithan TW. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Hum Mol Genet 1998; 7(4):755-61; PMID:9499431; http://dx.doi.org/10.1093/hmg/7.4.755
- Widrow RJ, Hansen RS, Kawame H, Gartler SM, Laird CD. Very late DNA replication in the human cell cycle. Proc Natl Acad Sci U S A 1998; 95(19):11246-50; PMID:9736721; http://dx.doi.org/10.1073/pnas.95.19.11246
- Hirano T. Chromosome Dynamics during Mitosis. Cold Spring Harb Perspect Biol 2015; 7(6):1–14; PMID:25722466; http://dx.doi.org/10.1101/cshperspect.a015792
- Tanaka K, Hirota T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim Biophys Acta 2016; 1866(1):64-75; PMID:27345585
- Bakhoum SF, Silkworth WT, Nardi IK, Nicholson JM, Compton DA, Cimini D. The mitotic origin of chromosomal instability. Curr Biol 2014; 24(4):R148-9; PMID:24556433; http://dx.doi.org/10.1016/j.cub.2014.01.019
- Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 2009; 11(6):761-8; PMID:19465921; http://dx.doi.org/10.1038/ncb1883
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 2009; 11(6):753-60; PMID:19465922; http://dx.doi.org/10.1038/ncb1882
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013; 494(7438):492-6; PMID:23446422; http://dx.doi.org/10.1038/nature11935
- Naim V, Rosselli F. The FANC pathway and mitosis: a replication legacy. Cell Cycle 2009; 8(18):2907-11; PMID:19729998; http://dx.doi.org/10.4161/cc.8.18.9538
- Chan KL, North PS, Hickson ID. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J 2007; 26(14):3397-409; PMID:17599064; http://dx.doi.org/10.1038/sj.emboj.7601777
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grøfte M, Chan KL, Hickson ID, Bartek J, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 2011; 13(3):243-53; PMID:21317883; http://dx.doi.org/10.1038/ncb2201
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 2011; 193(1):97-108; PMID:21444690; http://dx.doi.org/10.1083/jcb.201011083
- Baumann C, Körner R, Hofmann K, Nigg EA. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 2007; 128(1):101-14; PMID:17218258; http://dx.doi.org/10.1016/j.cell.2006.11.041
- Naim V, Wilhelm T, Debatisse M, Rosselli F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 2013; 15(8):1008-15; PMID:23811686; http://dx.doi.org/10.1038/ncb2793
- Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, Hickson ID. MUS81 promotes common fragile site expression. Nat Cell Biol 2013; 15(8):1001-7; PMID:23811685; http://dx.doi.org/10.1038/ncb2773
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. Replication stress activates DNA repair synthesis in mitosis. Nature 2015; 528(7581):286-90; PMID:26633632; http://dx.doi.org/10.1038/nature16139
- Pedersen RT, Kruse T, Nilsson J, Oestergaard VH, Lisby M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J Cell Biol 2015; 210(4):565-82; PMID:26283799; http://dx.doi.org/10.1083/jcb.201502107
- Wild P, Matos J. Cell cycle control of DNA joint molecule resolution. Curr Opin Cell Biol 2016; 40, 74-80; PMID:26970388; http://dx.doi.org/10.1016/j.ceb.2016.02.018
- Fragkos M, Ganier O, Coulombe P, Méchali M. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 2015; 16(6):360-74; PMID:25999062; http://dx.doi.org/10.1038/nrm4002
- Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, Hartford SA, Tye BK, Schimenti JC. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet 2007; 39(1):93-8; PMID:17143284; http://dx.doi.org/10.1038/ng1936
- Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, Shima N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol Cell 2011; 41(5):543-53; PMID:21362550; http://dx.doi.org/10.1016/j.molcel.2011.02.006
- Letessier A, Millot GA, Koundrioukoff S, Lachagès AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011; 470(7332):120-3; PMID:21258320; http://dx.doi.org/10.1038/nature09745
- Ozeri-Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, Kerem B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell 2011; 43(1):122-31; PMID:21726815; http://dx.doi.org/10.1016/j.molcel.2011.05.019
- Aparicio OM. Location, location, location: it's all in the timing for replication origins. Genes Dev 2013; 27(2):117-28; PMID:23348837; http://dx.doi.org/10.1101/gad.209999.112
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013; 155(5):1088-103; PMID:24267891; http://dx.doi.org/10.1016/j.cell.2013.10.043
- Magdalou I, Lopez BS, Pasero P, Lambert SA. The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol 2014; 30, 154-64; PMID:24818779; http://dx.doi.org/10.1016/j.semcdb.2014.04.035
- Blow JJ, Ge XQ, Jackson DA. How dormant origins promote complete genome replication. Trends Biochem Sci 2011; 36(8):405-14; PMID:21641805; http://dx.doi.org/10.1016/j.tibs.2011.05.002
- Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev 2007; 21(24):3331-41; PMID:18079179; http://dx.doi.org/10.1101/gad.457807
- Woodward AM, Göhler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol 2006; 173(5):673-83; PMID:16754955; http://dx.doi.org/10.1083/jcb.200602108
- Ibarra A, Schwob E, Méndez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci U S A 2008; 105(26):8956-61; PMID:18579778; http://dx.doi.org/10.1073/pnas.0803978105
- Anglana M, Apiou F, Bensimon A, Debatisse M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell 2003; 114(3):385-94; PMID:12914702; http://dx.doi.org/10.1016/S0092-8674(03)00569-5
- Rybak P, Waligórska A, Bujnowicz Ł, Hoang A, Dobrucki JW. Activation of new replication foci under conditions of replication stress. Cell Cycle 2015; 14(16):2634-47; PMID:26212617; http://dx.doi.org/10.1080/15384101.2015.1064566
- Cortez D. Preventing replication fork collapse to maintain genome integrity. DNA Repair (Amst) 2015; 32, 149-57; PMID:25957489; http://dx.doi.org/10.1016/j.dnarep.2015.04.026
- Branzei D, Foiani M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair (Amst) 2007; 6(7):994-1003; PMID:17382606; http://dx.doi.org/10.1016/j.dnarep.2007.02.018
- Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007; 6(7):953-66; PMID:17531546; http://dx.doi.org/10.1016/j.dnarep.2007.02.015
- Berti M, Vindigni A. Replication stress: getting back on track. Nat Struct Mol Biol 2016; 23(2):103-9; PMID:26840898; http://dx.doi.org/10.1038/nsmb.3163
- Lehmann AR. Replication of UV-damaged DNA: new insights into links between DNA polymerases, mutagenesis and human disease. Gene 2000; 253(1):1-12; PMID:10925197; http://dx.doi.org/10.1016/S0378-1119(00)00250-X
- Ward IM, Minn K, Chen J. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 2004; 279(11):9677-80; PMID:14742437; http://dx.doi.org/10.1074/jbc.C300554200
- Glover TW, Berger C, Coyle J, Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet 1984; 67(2):136-42; PMID:6430783; http://dx.doi.org/10.1007/BF00272988
- Byrnes JJ. Structural and functional properties of DNA polymerase delta from rabbit bone marrow. Mol Cell Biochem 1984; 62(1):13-24; PMID:6330522; http://dx.doi.org/10.1007/BF00230073
- Cheng CH, Kuchta RD. DNA polymerase epsilon: aphidicolin inhibition and the relationship between polymerase and exonuclease activity. Biochemistry 1993; 32(33):8568-74; PMID:8395209; http://dx.doi.org/10.1021/bi00084a025
- Takeuchi R, Oshige M, Uchida M, Ishikawa G, Takata K, Shimanouchi K, Kanai Y, Ruike T, Morioka H, Sakaguchi K. Purification of Drosophila DNA polymerase zeta by REV1 protein-affinity chromatography. Biochem J 2004; 382(Pt 2):535-43; PMID:15175013; http://dx.doi.org/10.1042/BJ20031833
- Poli J, Tsaponina O, Crabbé L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, Pasero P. dNTP pools determine fork progression and origin usage under replication stress. EMBO J 2012; 31(4):883-94; PMID:22234185; http://dx.doi.org/10.1038/emboj.2011.470
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37(4):492-502; PMID:20188668; http://dx.doi.org/10.1016/j.molcel.2010.01.021
- Aparicio JG, Viggiani CJ, Gibson DG, Aparicio OM. The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol Cell Biol 2004; 24(11):4769-80; PMID:15143171; http://dx.doi.org/10.1128/MCB.24.11.4769-4780.2004
- Bickmore WA, Carothers AD. Factors affecting the timing and imprinting of replication on a mammalian chromosome. J Cell Sci 1995; 108 ( Pt 8):2801-9; PMID:7593321
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 2006; 6(10):789-802; PMID:16990856; http://dx.doi.org/10.1038/nrc1977
- Lambert S, Carr AM. Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma 2013; 122(1-2):33-45; PMID:23446515; http://dx.doi.org/10.1007/s00412-013-0398-9
- Tarsounas M, Tijsterman M. Genomes and G-quadruplexes: for better or for worse. J Mol Biol 2013; 425(23):4782-9; PMID:24076189; http://dx.doi.org/10.1016/j.jmb.2013.09.026
- Mendoza O, Bourdoncle A, Boulé JB, Brosh RM Jr, Mergny JL. G-quadruplexes and helicases. Nucleic Acids Res 2016; 44(5):1989-2006; PMID:26883636; http://dx.doi.org/10.1093/nar/gkw079
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 2010; 11(3):208-19; PMID:20177396; http://dx.doi.org/10.1038/nrm2852
- Martinez P, Blasco MA. Replicating through telomeres: a means to an end. Trends Biochem Sci 2015; 40(9):504-15; PMID:26188776; http://dx.doi.org/10.1016/j.tibs.2015.06.003
- Zimmer J, Tacconi EM, Folio C, Badie S, Porru M, Klare K, Tumiati M, Markkanen E, Halder S, Ryan A, et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol Cell 2016; 61(3):449-60; PMID:26748828; http://dx.doi.org/10.1016/j.molcel.2015.12.004
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009; 138(1):90-103; PMID:19596237; http://dx.doi.org/10.1016/j.cell.2009.06.021
- Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 2012; 149(4):795-806; PMID:22579284; http://dx.doi.org/10.1016/j.cell.2012.03.030
- Aguilera A, Garcia-Muse T. Causes of genome instability. Annu Rev Genet 2013; 47, 1-32; PMID:23909437; http://dx.doi.org/10.1146/annurev-genet-111212-133232
- Lebofsky R, Bensimon A. DNA replication origin plasticity and perturbed fork progression in human inverted repeats. Mol Cell Biol 2005; 25(15):6789-97; PMID:16024811; http://dx.doi.org/10.1128/MCB.25.15.6789-6797.2005
- Akamatsu Y, Kobayashi T. The Human RNA Polymerase I Transcription Terminator Complex Acts as a Replication Fork Barrier That Coordinates the Progress of Replication with rRNA Transcription Activity. Mol Cell Biol 2015; 35(10):1871-81; PMID:25776556; http://dx.doi.org/10.1128/MCB.01521-14
- Brewer BJ, Fangman WL. A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 1988; 55(4):637-43; PMID:3052854; http://dx.doi.org/10.1016/0092-8674(88)90222-X
- Kobayashi T, Horiuchi T. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells 1996; 1(5):465-74; PMID:9078378; http://dx.doi.org/10.1046/j.1365-2443.1996.d01-256.x
- Linskens MH, Huberman JA. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol Cell Biol 1988; 8(11):4927-35; PMID:3062373; http://dx.doi.org/10.1128/MCB.8.11.4927
- Lopez-estrano C, Schvartzman JB, Krimer DB, Hernández P. Co-localization of polar replication fork barriers and rRNA transcription terminators in mouse rDNA. J Mol Biol 1998; 277(2):249-56; PMID:9514756; http://dx.doi.org/10.1006/jmbi.1997.1607
- Gerber JK, Gögel E, Berger C, Wallisch M, Müller F, Grummt I, Grummt F. Termination of mammalian rDNA replication: polar arrest of replication fork movement by transcription termination factor TTF-I. Cell 1997; 90(3):559-67; PMID:9267035; http://dx.doi.org/10.1016/S0092-8674(00)80515-2
- Probst AV, Almouzni G. Pericentric heterochromatin: dynamic organization during early development in mammals. Differentiation 2008; 76(1):15-23; PMID:17825083; http://dx.doi.org/10.1111/j.1432-0436.2007.00220.x
- Porter AC, Farr CJ. Topoisomerase II: untangling its contribution at the centromere. Chromosome Res 2004; 12(6):569-83; PMID:15289664; http://dx.doi.org/10.1023/B:CHRO.0000036608.91085.d1
- Rouzeau S, Cordelières FP, Buhagiar-Labarchède G, Hurbain I, Onclercq-Delic R, Gemble S, Magnaghi-Jaulin L, Jaulin C, Amor-Guéret M. Bloom's syndrome and PICH helicases cooperate with topoisomerase IIalpha in centromere disjunction before anaphase. PLoS One 2012; 7(4):e33905; PMID:22563370; http://dx.doi.org/10.1371/journal.pone.0033905
- Azvolinsky A, Dunaway S, Torres JZ, Bessler JB, Zakian VA. The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev 2006; 20(22):3104-16; PMID:17114583; http://dx.doi.org/10.1101/gad.1478906
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434(7035):907-13; PMID:15829965; http://dx.doi.org/10.1038/nature03485
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319(5868):1352-5; PMID:18323444; http://dx.doi.org/10.1126/science.1140735
- Maya-Mendoza A, Ostrakova J, Kosar M, Hall A, Duskova P, Mistrik M, Merchut-Maya JM, Hodny Z, Bartkova J, Christensen C, et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol Oncol 2015; 9(3):601-16; PMID:25435281; http://dx.doi.org/10.1016/j.molonc.2014.11.001
- Tsantoulis PK, Kotsinas A, Sfikakis PP, Evangelou K, Sideridou M, Levy B, Mo L, Kittas C, Wu XR, Papavassiliou AG, et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008; 27(23):3256-64
- Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434(7035):864-70; PMID:15829956; http://dx.doi.org/10.1038/nature03482
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444(7119):633-7; PMID:17136093; http://dx.doi.org/10.1038/nature05268
- Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, Fumagalli M, Di Micco R, Mirani N, Gurung RL. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J 2012; 31(13):2839-51; PMID:22569128; http://dx.doi.org/10.1038/emboj.2012.132
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre′ M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444(7119):638-42; PMID:17136094; http://dx.doi.org/10.1038/nature05327
- Hills SA, Diffley JF. DNA replication and oncogene-induced replicative stress. Curr Biol 2014; 24(10):R435-44; PMID:24845676; http://dx.doi.org/10.1016/j.cub.2014.04.012
- Ekholm-Reed S, Méndez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol 2004; 165(6):789-800; PMID:15197178; http://dx.doi.org/10.1083/jcb.200404092
- Jones RM, Mortusewicz O, Afzal I, Lorvellec M, García P, Helleday T, Petermann E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013; 32(32):3744-53; PMID:22945645; http://dx.doi.org/10.1038/onc.2012.387
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011; 145(3):435-46; PMID:21529715; http://dx.doi.org/10.1016/j.cell.2011.03.044
- Xie M, Yen Y, Owonikoko TK, Ramalingam SS, Khuri FR, Curran WJ, Doetsch PW, Deng X. Bcl2 induces DNA replication stress by inhibiting ribonucleotide reductase. Cancer Res 2014; 74(1):212-23; PMID:24197132; http://dx.doi.org/10.1158/0008-5472.CAN-13-1536-T
- Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res 2007; 35(22):7545-56; PMID:18055498; http://dx.doi.org/10.1093/nar/gkm1059
- Ogrunc M, Di Micco R, Liontos M, Bombardelli L, Mione M, Fumagalli M, Gorgoulis VG, d'Adda di Fagagna F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ 2014; 21(6):998-1012; PMID:24583638; http://dx.doi.org/10.1038/cdd.2014.16
- Wilhelm T, Ragu S, Magdalou I, Machon C, Dardillac E, Técher H, Guitton J, Debatisse M, Lopez BS. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet 2016; 12(5):e1006007; PMID:27135742; http://dx.doi.org/10.1371/journal.pgen.1006007
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119(7):941-53; PMID:15620353; http://dx.doi.org/10.1016/j.cell.2004.12.012
- Brooks PJ, Theruvathu JA. DNA adducts from acetaldehyde: implications for alcohol-related carcinogenesis. Alcohol 2005; 35(3):187-93; PMID:16054980; http://dx.doi.org/10.1016/j.alcohol.2005.03.009
- Garaycoechea JI, Patel KJ. Why does the bone marrow fail in Fanconi anemia? Blood 2014; 123(1):26-34; PMID:24200684; http://dx.doi.org/10.1182/blood-2013-09-427740
- McClendon AK, Rodriguez AC, Osheroff N. Human topoisomerase IIalpha rapidly relaxes positively supercoiled DNA: implications for enzyme action ahead of replication forks. J Biol Chem 2005; 280(47):39337-45; PMID:16188892; http://dx.doi.org/10.1074/jbc.M503320200
- Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell 2007; 128(4):721-33; PMID:17320509; http://dx.doi.org/10.1016/j.cell.2007.01.030
- Clemente-Ruiz M, Prado F. Chromatin assembly controls replication fork stability. EMBO Rep 2009; 10(7):790-6; PMID:19465889; http://dx.doi.org/10.1038/embor.2009.67
- Mejlvang J, et al. New histone supply regulates replication fork speed and PCNA unloading. J Cell Biol 2014; 204(1):29-43; http://dx.doi.org/10.1083/jcb.201305017
- Nikolov I, Taddei A. Linking replication stress with heterochromatin formation. Chromosoma 2016; 125(3):523-33; http://dx.doi.org/10.1007/s00412-015-0545-6
- Tabancay AP, Jr., Forsburg SL. Eukaryotic DNA replication in a chromatin context. Curr Top Dev Biol 76, 129-84; PMID:17118266
- Wei X, Samarabandu J, Devdhar RS, Siegel AJ, Acharya R, Berezney R. (1998) Segregation of transcription and replication sites into higher order domains. Science 2006; 281(5382):1502-6; PMID:9727975; http://dx.doi.org/10.1126/science.281.5382.1502
- Gottipati P, Cassel TN, Savolainen L, Helleday T. Transcription-associated recombination is dependent on replication in Mammalian cells. Mol Cell Biol 2008; 28(1):154-64; PMID:17967877; http://dx.doi.org/10.1128/MCB.00816-07
- Poli J, Gerhold CB, Tosi A, Hustedt N, Seeber A, Sack R, Herzog F, Pasero P, Shimada K, Hopfner KP, et al. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev 2016; 30(3):337-54; PMID:26798134; http://dx.doi.org/10.1101/gad.273813.115
- Lafon A, Taranum S, Pietrocola F, Dingli F, Loew D, Brahma S, Bartholomew B, Papamichos-Chronakis M. INO80 Chromatin Remodeler Facilitates Release of RNA Polymerase II from Chromatin for Ubiquitin-Mediated Proteasomal Degradation. Mol Cell 2015; 60(5):784-96; PMID:26656161; http://dx.doi.org/10.1016/j.molcel.2015.10.028
- Poveda AM, Le Clech M, Pasero P. Transcription and replication: breaking the rules of the road causes genomic instability. Transcription 2010; 1(2):99-102; PMID:21326900; http://dx.doi.org/10.4161/trns.1.2.12665
- Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet 2015; 16(10):583-97; PMID:26370899; http://dx.doi.org/10.1038/nrg3961
- Sollier J, Stork CT, García-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 2014; 56(6):777-85; PMID:25435140; http://dx.doi.org/10.1016/j.molcel.2014.10.020
- Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M, et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012; 151(4):835-46; PMID:23141540; http://dx.doi.org/10.1016/j.cell.2012.09.041
- Li X, Manley JL. Cotranscriptional processes and their influence on genome stability. Genes Dev 2006; 20(14):1838-47; PMID:16847344; http://dx.doi.org/10.1101/gad.1438306
- Gomez-Gonzalez B, García-Rubio M, Bermejo R, Gaillard H, Shirahige K, Marín A, Foiani M, Aguilera A. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J 2011; 30(15):3106-19; PMID:21701562; http://dx.doi.org/10.1038/emboj.2011.206
- Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev 2012; 26(2):163-75; PMID:22279048; http://dx.doi.org/10.1101/gad.179721.111
- Wahba L, Gore SK, Koshland D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. Elife 2, e00505; PMID:23795288; http://dx.doi.org/10.7554/eLife.00505
- Ginno PA, Lim YW, Lott PL, Korf I, Chédin F. ( 2013) GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res 2013; 23(10):1590-600; PMID:23868195; http://dx.doi.org/10.1101/gr.158436.113
- Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell 2012; 46(2):115-24; PMID:22541554; http://dx.doi.org/10.1016/j.molcel.2012.04.009
- Tuduri S, Crabbé L, Conti C, Tourrière H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 2009; 11(11):1315-24; PMID:19838172; http://dx.doi.org/10.1038/ncb1984
- Bermejo R, Capra T, Gonzalez-Huici V, Fachinetti D, Cocito A, Natoli G, Katou Y, Mori H, Kurokawa K, Shirahige K, et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 2009; 138(5):870-84; PMID:19737516; http://dx.doi.org/10.1016/j.cell.2009.06.022
- Bermejo R, Capra T, Jossen R, Colosio A, Frattini C, Carotenuto W, Cocito A, Doksani Y, Klein H, Gómez-González B, et al. The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell 2011; 146(2):233-46; PMID:21784245; http://dx.doi.org/10.1016/j.cell.2011.06.033
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet 41, 169-92; PMID:17608616; http://dx.doi.org/10.1146/annurev.genet.41.042007.165900
- Sutherland GR ( 1977) Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science 2007; 197(4300):265-6; PMID:877551; http://dx.doi.org/10.1126/science.877551
- Sutherland GR. The role of nucleotides in human fragile site expression. Mutat Res 1988; 200(1-2):207-13; PMID:3292907; http://dx.doi.org/10.1016/0027-5107(88)90084-X
- Jiang Y, Lucas I, Young DJ, Davis EM, Karrison T, Rest JS, Le Beau MM. Common fragile sites are characterized by histone hypoacetylation. Hum Mol Genet 2009; 18(23):4501-12; PMID:19717471; http://dx.doi.org/10.1093/hmg/ddp410
- Becker NA, Thorland EC, Denison SR, Phillips LA, Smith DI. Evidence that instability within the FRA3B region extends four megabases. Oncogene 2002; 21(57):8713-22; PMID:12483524; http://dx.doi.org/10.1038/sj.onc.1205950
- Smith DI, Zhu Y, McAvoy S, Kuhn R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett 2006; 232(1):48-57; PMID:16221525; http://dx.doi.org/10.1016/j.canlet.2005.06.049
- Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O. Common fragile sites: mechanisms of instability revisited. Trends Genet 2012; 28(1):22-32; PMID:22094264; http://dx.doi.org/10.1016/j.tig.2011.10.003
- Le Tallec B, Koundrioukoff S, Wilhelm T, Letessier A, Brison O, Debatisse M. Updating the mechanisms of common fragile site instability: how to reconcile the different views? Cell Mol Life Sci 2014; 71(23):4489-94; PMID:25248392; http://dx.doi.org/10.1007/s00018-014-1720-2
- Ozeri-Galai E, Tur-Sinai M, Bester AC, Kerem B. Interplay between genetic and epigenetic factors governs common fragile site instability in cancer. Cell Mol Life Sci 2014; 71(23):4495-506; PMID:25297918; http://dx.doi.org/10.1007/s00018-014-1719-8
- Sarni D, Kerem B. The complex nature of fragile site plasticity and its importance in cancer. Curr Opin Cell Biol 40, 131-6; PMID:27062332; http://dx.doi.org/10.1016/j.ceb.2016.03.017
- Miotto B, Ji Z, Struhl K. ( 2016) Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc Natl Acad Sci U S A 2016; 113(33):E4810-9; PMID:27436900; http://dx.doi.org/10.1073/pnas.1609060113
- Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 2011; 44(6):966-77; PMID:22195969; http://dx.doi.org/10.1016/j.molcel.2011.10.013
- Wilson TE, Arlt MF, Park SH, Rajendran S, Paulsen M, Ljungman M, Glover TW. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res 2015; 25(2):189-200; PMID:25373142; http://dx.doi.org/10.1101/gr.177121.114
- Gros J, Kumar C, Lynch G, Yadav T, Whitehouse I, Remus D. Post-licensing Specification of Eukaryotic Replication Origins by Facilitated Mcm2-7 Sliding along DNA. Mol Cell 2015; 60(5):797-807; PMID:26656162; http://dx.doi.org/10.1016/j.molcel.2015.10.022
- Snyder M, Sapolsky RJ, Davis RW. Transcription interferes with elements important for chromosome maintenance in Saccharomyces cerevisiae. Mol Cell Biol 1988; 8(5):2184-94; PMID:3290652; http://dx.doi.org/10.1128/MCB.8.5.2184
- Looke M, Reimand J, Sedman T, Sedman J, Järvinen L, Värv S, Peil K, Kristjuhan K, Vilo J, Kristjuhan A. Relicensing of transcriptionally inactivated replication origins in budding yeast. J Biol Chem 2010; 285(51):40004-11; PMID:20962350; http://dx.doi.org/10.1074/jbc.M110.148924
- Miron K, Golan-Lev T, Dvir R, Ben-David E, Kerem B. Oncogenes create a unique landscape of fragile sites. Nat Commun 2015; 6, 7094; PMID:25959793; http://dx.doi.org/10.1038/ncomms8094
- Miron K, Kerem B. To break or not to break - context matters. Mol Cell Oncol 2016; 3(1):e1072657; PMID:27308576; http://dx.doi.org/10.1080/23723556.2015.1072657
- Barlow JH, Faryabi RB, Callén E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013; 152(3):620-32; PMID:23352430; http://dx.doi.org/10.1016/j.cell.2013.01.006
- Koundrioukoff S, Carignon S, Técher H, Letessier A, Brison O, Debatisse M. Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet 2013; 9(7):e1003643; PMID:23874235; http://dx.doi.org/10.1371/journal.pgen.1003643
- Magiera MM, Gueydon E, Schwob E. DNA replication and spindle checkpoints cooperate during S phase to delay mitosis and preserve genome integrity. J Cell Biol 2014; 204(2):165-75; PMID:24421333; http://dx.doi.org/10.1083/jcb.201306023
- Mankouri HW, Huttner D, Hickson ID. How unfinished business from S-phase affects mitosis and beyond. EMBO J 2013; 32(20):2661-71; PMID:24065128; http://dx.doi.org/10.1038/emboj.2013.211
- Hossain M, Stillman B. Meier-Gorlin syndrome mutations disrupt an Orc1 CDK inhibitory domain and cause centrosome reduplication. Genes Dev 2012; 26(16):1797-810; PMID:22855792; http://dx.doi.org/10.1101/gad.197178.112
- Laulier C, Cheng A, Stark JM. The relative efficiency of homology-directed repair has distinct effects on proper anaphase chromosome separation. Nucleic Acids Res 2011; 39(14):5935-44; PMID:21459848; http://dx.doi.org/10.1093/nar/gkr187
- Wilhelm T, Magdalou I, Barascu A, Técher H, Debatisse M, Lopez BS. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc Natl Acad Sci U S A 2014; 111(2):763-8; PMID:24347643; http://dx.doi.org/10.1073/pnas.1311520111
- Gelot C, Magdalou I, Lopez BS. Replication stress in Mammalian cells and its consequences for mitosis. Genes (Basel) 2015; 6(2):267-98; PMID:26010955
- Kramer A, Mailand N, Lukas C, Syljuåsen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol 2004; 6(9):884-91; PMID:15311285; http://dx.doi.org/10.1038/ncb1165
- Chan KL, Hickson ID. New insights into the formation and resolution of ultra-fine anaphase bridges. Semin Cell Dev Biol 2011; 22(8):906-12; PMID:21782962; http://dx.doi.org/10.1016/j.semcdb.2011.07.001
- Biebricher A, Hirano S, Enzlin JH, Wiechens N, Streicher WW, Huttner D, Wang LH, Nigg EA, Owen-Hughes T, Liu Y, et al. PICH: a DNA translocase specially adapted for processing anaphase bridge DNA. Mol Cell 2013; 51(5):691-701; PMID:23973328; http://dx.doi.org/10.1016/j.molcel.2013.07.016
- Germann SM, Schramke V, Pedersen RT, Gallina I, Eckert-Boulet N, Oestergaard VH, Lisby M. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J Cell Biol 2014; 204(1):45-59; PMID:24379413; http://dx.doi.org/10.1083/jcb.201305157
- Liu Y, Nielsen CF, Yao Q, Hickson ID. The origins and processing of ultra fine anaphase DNA bridges. Curr Opin Genet Dev 2014; 26, 1-5; PMID:24795279; http://dx.doi.org/10.1016/j.gde.2014.03.003
- Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet 2009; 43, 525-58; PMID:19886810; http://dx.doi.org/10.1146/annurev-genet-102108-134233
- Wang LH, Schwarzbraun T, Speicher MR, Nigg EA. Persistence of DNA threads in human anaphase cells suggests late completion of sister chromatid decatenation. Chromosoma 2008; 117(2):123-35; PMID:17989990; http://dx.doi.org/10.1007/s00412-007-0131-7
- Wang LH, Mayer B, Stemmann O, Nigg EA. Centromere DNA decatenation depends on cohesin removal and is required for mammalian cell division. J Cell Sci 2010; 123(Pt 5):806-13; PMID:20144989; http://dx.doi.org/10.1242/jcs.058255
- Barefield C, Karlseder J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res 2012; 40(15):7358-67; PMID:22576367; http://dx.doi.org/10.1093/nar/gks407
- d'Alcontres MS, Palacios JA, Mejias D, Blasco MA. TopoIIalpha prevents telomere fragility and formation of ultra thin DNA bridges during mitosis through TRF1-dependent binding to telomeres. Cell Cycle 2014; 13(9):1463-81; PMID:24626180; http://dx.doi.org/10.4161/cc.28419
- Nera B, Huang HS, Lai T, Xu L. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat Commun 2015; 6, 10132; PMID:26640040; http://dx.doi.org/10.1038/ncomms10132
- Zaaijer S, Shaikh N, Nageshan RK, Cooper JP. Rif1 Regulates the Fate of DNA Entanglements during Mitosis. Cell Rep 2016; 16(1):148-60; PMID:27320927; http://dx.doi.org/10.1016/j.celrep.2016.05.077
- Sofueva S, Osman F, Lorenz A, Steinacher R, Castagnetti S, Ledesma J, Whitby MC. Ultrafine anaphase bridges, broken DNA and illegitimate recombination induced by a replication fork barrier. Nucleic Acids Res 2011; 39(15):6568-84; PMID:21576223; http://dx.doi.org/10.1093/nar/gkr340
- Martinez P, Thanasoula M, Muñoz P, Liao C, Tejera A, McNees C, Flores JM, Fernández-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev 2009; 23(17):2060-75; PMID:19679647; http://dx.doi.org/10.1101/gad.543509
- Ke Y, Huh JW, Warrington R, Li B, Wu N, Leng M, Zhang J, Ball HL, Li B, Yu H. PICH and BLM limit histone association with anaphase centromeric DNA threads and promote their resolution. EMBO J 2011; 30(16):3309-21; PMID:21743438; http://dx.doi.org/10.1038/emboj.2011.226
- Kaulich M, Cubizolles F, Nigg EA. On the regulation, function, and localization of the DNA-dependent ATPase PICH. Chromosoma 2012; 121(4):395-408; PMID:22527115; http://dx.doi.org/10.1007/s00412-012-0370-0
- Nielsen CF, Huttner D, Bizard AH, Hirano S, Li TN, Palmai-Pallag T, Bjerregaard VA, Liu Y, Nigg EA, Wang LH. PICH promotes sister chromatid disjunction and co-operates with topoisomerase II in mitosis. Nat Commun 2015; 6, 8962; PMID:26643143; http://dx.doi.org/10.1038/ncomms9962
- Nielsen CF, Hickson ID. PICH promotes mitotic chromosome segregation: Identification of a novel role in rDNA disjunction. Cell Cycle 2016; 15(20):2704-11; PMID:27565185; http://dx.doi.org/10.1080/15384101.2016.1222336
- Broderick R, Nieminuszczy J, Blackford AN, Winczura A, Niedzwiedz W. TOPBP1 recruits TOP2A to ultra-fine anaphase bridges to aid in their resolution. Nat Commun 2015; 6, 6572; PMID:25762097; http://dx.doi.org/10.1038/ncomms7572
- Vinciguerra P, Godinho SA, Parmar K, Pellman D, D'Andrea AD. Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J Clin Invest 2010; 120(11):3834-42; PMID:20921626; http://dx.doi.org/10.1172/JCI43391
- Mattarocci S, Hafner L, Lezaja A, Shyian M, Shore D. Rif1: A Conserved Regulator of DNA Replication and Repair Hijacked by Telomeres in Yeasts. Front Genet 2016; 7, 45; PMID:27066066; http://dx.doi.org/10.3389/fgene.2016.00045
- Hengeveld RC, de Boer HR, Schoonen PM, de Vries EG, Lens SM, van Vugt MA. Rif1 Is Required for Resolution of Ultrafine DNA Bridges in Anaphase to Ensure Genomic Stability. Dev Cell 2015; 34(4):466-74; PMID:26256213; http://dx.doi.org/10.1016/j.devcel.2015.06.014
- Rodrigue A, Coulombe Y, Jacquet K, Gagné JP, Roques C, Gobeil S, Poirier G, Masson JY. The RAD51 paralogs ensure cellular protection against mitotic defects and aneuploidy. J Cell Sci 2013; 126(Pt 1):348-59; PMID:23108668; http://dx.doi.org/10.1242/jcs.114595
- Galgoczy DJ, Toczyski DP. Checkpoint adaptation precedes spontaneous and damage-induced genomic instability in yeast. Mol Cell Biol 2001; 21(5):1710-8; PMID:11238908; http://dx.doi.org/10.1128/MCB.21.5.1710-1718.2001
- Syljuasen RG, Jensen S, Bartek J, Lukas J. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res 2006; 66(21):10253-7; PMID:17079442; http://dx.doi.org/10.1158/0008-5472.CAN-06-2144
- Yoo HY, Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 2004; 117(5):575-88; PMID:15163406; http://dx.doi.org/10.1016/S0092-8674(04)00417-9
- Eykelenboom JK, Harte EC, Canavan L, Pastor-Peidro A, Calvo-Asensio I, Llorens-Agost M, Lowndes NF. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep 2013; 5(4):1095-107; PMID:24268773; http://dx.doi.org/10.1016/j.celrep.2013.10.027
- Torres-Rosell J, De Piccoli G, Cordon-Preciado V, Farmer S, Jarmuz A, Machin F, Pasero P, Lisby M, Haber JE, Aragón L. Anaphase onset before complete DNA replication with intact checkpoint responses. Science 2007; 315(5817):1411-5; PMID:17347440; http://dx.doi.org/10.1126/science.1134025
- Bogliolo M, Surralles J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev 2015; 33, 32-40; PMID:26254775; http://dx.doi.org/10.1016/j.gde.2015.07.002
- Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 2016; 17(6):337-49; PMID:27145721; http://dx.doi.org/10.1038/nrm.2016.48
- Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, Dubois d'Enghien C, Bluteau O, Cuccuini W, Gachet S, Peffault de Latour R, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest 2016; 126(9):3580-4; PMID:27500492; http://dx.doi.org/10.1172/JCI88010
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22(1):106-16; PMID:22789542; http://dx.doi.org/10.1016/j.ccr.2012.05.015
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Décaillet C, Gari K, Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 2013; 51(5):678-90; PMID:23993743; http://dx.doi.org/10.1016/j.molcel.2013.07.023
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 2013; 288(44):31458-67; PMID:24047897; http://dx.doi.org/10.1074/jbc.M113.511337
- Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ 3rd, Rothenberg E, Jallepalli PV, Huang TT. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell 2015; 58(2):323-38; PMID:25843623; http://dx.doi.org/10.1016/j.molcel.2015.02.031
- Shigechi T, Tomida J, Sato K, Kobayashi M, Eykelenboom JK, Pessina F, Zhang Y, Uchida E, Ishiai M, Lowndes NF, et al. ATR-ATRIP kinase complex triggers activation of the Fanconi anemia DNA repair pathway. Cancer Res 2012; 72(5):1149-56; PMID:22258451; http://dx.doi.org/10.1158/0008-5472.CAN-11-2904
- Tomida J, Itaya A, Shigechi T, Unno J, Uchida E, Ikura M, Masuda Y, Matsuda S, Adachi J, Kobayashi M, et al. A novel interplay between the Fanconi anemia core complex and ATR-ATRIP kinase during DNA cross-link repair. Nucleic Acids Res 2013; 41(14):6930-41; PMID:23723247; http://dx.doi.org/10.1093/nar/gkt467
- Stirling PC, Hieter P. Canonical DNA Repair Pathways Influence R-Loop-Driven Genome Instability. J Mol Biol 2016; in press; PMID:27452366
- Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell 2015; 57(4):636-47; PMID:25699710; http://dx.doi.org/10.1016/j.molcel.2015.01.011
- Bhatia V, Barroso SI, García-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014; 511(7509):362-5; PMID:24896180; http://dx.doi.org/10.1038/nature13374
- Garcia-Rubio ML, Pérez-Calero C, Barroso SI, Tumini E, Herrera-Moyano E, Rosado IV, Aguilera A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet 2015; 11(11):e1005674; PMID:26584049; http://dx.doi.org/10.1371/journal.pgen.1005674
- Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell 2015; 60(3):351-61; PMID:26593718; http://dx.doi.org/10.1016/j.molcel.2015.09.012
- Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet 2005; 14(5):693-701; PMID:15661754; http://dx.doi.org/10.1093/hmg/ddi065
- Madireddy A, Kosiyatrakul ST, Boisvert RA, Herrera-Moyano E, García-Rubio ML, Gerhardt J, Vuono EA, Owen N, Yan Z, Olson S, et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol Cell 2016; 64(2):388-404; PMID:27768874; http://dx.doi.org/10.1016/j.molcel.2016.09.017
- Pichierri P, Franchitto A, Rosselli F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J 2004; 23(15):3154-63; PMID:15257300; http://dx.doi.org/10.1038/sj.emboj.7600277
- Chaudhury I, Sareen A, Raghunandan M, Sobeck A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res 2013; 41(13):6444-59; PMID:23658231; http://dx.doi.org/10.1093/nar/gkt348
- Deans AJ, West SC. FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia. Mol Cell 2009; 36(6):943-53; PMID:20064461; http://dx.doi.org/10.1016/j.molcel.2009.12.006
- Ciccia A, McDonald N, West SC. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem 2008; 77, 259-87; PMID:18518821; http://dx.doi.org/10.1146/annurev.biochem.77.070306.102408
- Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, Kanaar R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol 2007; 14(11):1096-104; PMID:17934473; http://dx.doi.org/10.1038/nsmb1313
- Guervilly JH, Takedachi A, Naim V, Scaglione S, Chawhan C, Lovera Y, Despras E, Kuraoka I, Kannouche P, Rosselli F, et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol Cell 2015; 57(1):123-37; PMID:25533188; http://dx.doi.org/10.1016/j.molcel.2014.11.014
- Ouyang J, Garner E, Hallet A, Nguyen HD, Rickman KA, Gill G, Smogorzewska A, Zou L. Noncovalent interactions with SUMO and ubiquitin orchestrate distinct functions of the SLX4 complex in genome maintenance. Mol Cell 2015; 57(1):108-22; PMID:25533185; http://dx.doi.org/10.1016/j.molcel.2014.11.015
- Kim Y. Nuclease delivery: versatile functions of SLX4/FANCP in genome maintenance. Mol Cells 2014; 37(8):569-74; http://dx.doi.org/10.14348/molcells.2014.0118
- Bhat A, et al. Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res 2013; 41(4):2328-39; http://dx.doi.org/10.1093/nar/gks1442
- Luebben SW, Kawabata T, Johnson CS, O'Sullivan MG, Shima N. A concomitant loss of dormant origins and FANCC exacerbates genome instability by impairing DNA replication fork progression. Nucleic Acids Res 2014; 42(9):5605-15; PMID:24589582; http://dx.doi.org/10.1093/nar/gku170
- Gemble S, Ahuja A, Buhagiar-Labarchède G, Onclercq-Delic R, Dairou J, Biard DS, Lambert S, Lopes M, Amor-Guéret M. Pyrimidine Pool Disequilibrium Induced by a Cytidine Deaminase Deficiency Inhibits PARP-1 Activity, Leading to the Under Replication of DNA. PLoS Genet 2015; 11(7):e1005384; PMID:26181065; http://dx.doi.org/10.1371/journal.pgen.1005384
- Malkova A, Ira G. Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev 2013; 23(3):271-9; PMID:23790415; http://dx.doi.org/10.1016/j.gde.2013.05.007
- Anand RP, Lovett ST, Haber JE. Break-induced DNA replication. Cold Spring Harb Perspect Biol 2013; 5(12):a010397; PMID:23881940; http://dx.doi.org/10.1101/cshperspect.a010397
- Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007; 448(7155):820-3; PMID:17671506; http://dx.doi.org/10.1038/nature06047
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014; 343(6166):88-91; PMID:24310611; http://dx.doi.org/10.1126/science.1243211
- Lahkim Bennani-Belhaj K, Rouzeau S, Buhagiar-Labarchède G, Chabosseau P, Onclercq-Delic R, Bayart E, Cordelières F, Couturier J, Amor-Guéret M. The Bloom syndrome protein limits the lethality associated with RAD51 deficiency. Mol Cancer Res 2010; 8(3):385-94; PMID:20215422; http://dx.doi.org/10.1158/1541-7786.MCR-09-0534
- Heijink AM, Krajewska M, van Vugt MA. The DNA damage response during mitosis. Mutat Res 2013; 750(1-2):45-55; PMID:23880065; http://dx.doi.org/10.1016/j.mrfmmm.2013.07.003
- Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J Cell Biol 2010; 190(2):197-207; PMID:20660628; http://dx.doi.org/10.1083/jcb.200911156
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014; 344(6180):189-93; PMID:24652939; http://dx.doi.org/10.1126/science.1248024
- Moriel-Carretero M, Aguilera A. A postincision-deficient TFIIH causes replication fork breakage and uncovers alternative Rad51- or Pol32-mediated restart mechanisms. Mol Cell 2010; 37(5):690-701; PMID:20227372; http://dx.doi.org/10.1016/j.molcel.2010.02.008
- Matos J, West SC. Holliday junction resolution: regulation in space and time. DNA Repair (Amst) 2014; 19, 176-81; PMID:24767945; http://dx.doi.org/10.1016/j.dnarep.2014.03.013
- Neelsen KJ, Zanini IM, Herrador R, Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol 2013; 200(6):699-708; PMID:23479741; http://dx.doi.org/10.1083/jcb.201212058
- Pardo B, Aguilera A. Complex chromosomal rearrangements mediated by break-induced replication involve structure-selective endonucleases. PLoS Genet 2012; 8(9):e1002979; PMID:23071463; http://dx.doi.org/10.1371/journal.pgen.1002979
- Ho CK, Mazón G, Lam AF, Symington LS. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell 2010; 40(6):988-1000; PMID:21172663; http://dx.doi.org/10.1016/j.molcel.2010.11.016
- Mayle R, et al. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 2015; 349(6249):742-7
- Guervilly JH, Gaillard PH. SLX4 gains weight with SUMO in genome maintenance. Mol Cell Oncol 2016; 3(2):e1008297; PMID:27308578; http://dx.doi.org/10.1080/23723556.2015.1008297
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009; 326(5960):1698-701; PMID:19965384; http://dx.doi.org/10.1126/science.1182372
- Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 2009; 73(1):134-54; PMID:19258535; http://dx.doi.org/10.1128/MMBR.00034-08
- Friedberg EC. Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol 2005; 6(12):943-53; PMID:16341080; http://dx.doi.org/10.1038/nrm1781
- Branzei D, Szakal B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair (Amst) 2016; 44, 68-75; PMID:27236213; http://dx.doi.org/10.1016/j.dnarep.2016.05.008
- Goodman MF, Woodgate R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol 2013; 5(10):a010363; PMID:23838442; http://dx.doi.org/10.1101/cshperspect.a010363
- Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 2004; 14(4):491-500; PMID:15149598; http://dx.doi.org/10.1016/S1097-2765(04)00259-X
- Chen X, Bosques L, Sung P, Kupfer GM. A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene 2016; 35(1):22-34; PMID:25893307; http://dx.doi.org/10.1038/onc.2015.68
- Gallina I, Christiansen SK, Pedersen RT, Lisby M, Oestergaard VH. TopBP1-mediated DNA processing during mitosis. Cell Cycle 2016; 15(2):176-83; PMID:26701150; http://dx.doi.org/10.1080/15384101.2015.1128595
- Boyer AS, Grgurevic S, Cazaux C, Hoffmann JS. The human specialized DNA polymerases and non-B DNA: vital relationships to preserve genome integrity. J Mol Biol 2013; 425(23):4767-81; PMID:24095858; http://dx.doi.org/10.1016/j.jmb.2013.09.022
- Baranovskiy AG, Lada AG, Siebler HM, Zhang Y, Pavlov YI, Tahirov TH. DNA polymerase delta and zeta switch by sharing accessory subunits of DNA polymerase delta. J Biol Chem 2012; 287(21):17281-7; PMID:22465957; http://dx.doi.org/10.1074/jbc.M112.351122
- Johnson RE, Prakash L, Prakash S. Pol31 and Pol32 subunits of yeast DNA polymerase delta are also essential subunits of DNA polymerase zeta. Proc Natl Acad Sci U S A 2012; 109(31):12455-60; PMID:22711820; http://dx.doi.org/10.1073/pnas.1206052109
- Makarova AV, Stodola JL, Burgers PM. A four-subunit DNA polymerase zeta complex containing Pol delta accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res 2012; 40(22):11618-26; PMID:23066099; http://dx.doi.org/10.1093/nar/gks948
- Sakofsky CJ, Ayyar S, Deem AK, Chung WH, Ira G, Malkova A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol Cell 2015; 60(6):860-72; PMID:26669261; http://dx.doi.org/10.1016/j.molcel.2015.10.041
- Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell 2002; 111(6):779-89; PMID:12526805; http://dx.doi.org/10.1016/S0092-8674(02)01113-3
- Hu CM, Chang ZF. Mitotic control of dTTP pool: a necessity or coincidence? J Biomed Sci 2007; 14(4):491-7; PMID:17525869; http://dx.doi.org/10.1007/s11373-007-9175-1
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene 2005; 24(17):2899-908; PMID:15838523; http://dx.doi.org/10.1038/sj.onc.1208615
- Yaswen P, Campisi J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell 2007; 128(2):233-4; PMID:17254959; http://dx.doi.org/10.1016/j.cell.2007.01.005
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011; 12(6):385-92; PMID:21527953; http://dx.doi.org/10.1038/nrm3115
- Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer 2007; 7(12):911-24; PMID:18004399; http://dx.doi.org/10.1038/nrc2249
- Fragkos M, Beard P. Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS One 2011; 6(8):e22946; PMID:21853057; http://dx.doi.org/10.1371/journal.pone.0022946
- Moreno A, Carrington JT, Albergante L, Al Mamun M, Haagensen EJ, Komseli ES, Gorgoulis VG, Newman TJ, Blow JJ. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc Natl Acad Sci U S A 2016; 113(39):E5757-64; PMID:27516545; http://dx.doi.org/10.1073/pnas.1603252113
- Ganem NJ, Pellman D. Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol 2012; 199(6):871-81; PMID:23229895; http://dx.doi.org/10.1083/jcb.201210040
- Zimmermann M, de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol 2014; 24(2):108-17; PMID:24094932; http://dx.doi.org/10.1016/j.tcb.2013.09.003
- Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011; 333(6051):1895-8; PMID:21960636; http://dx.doi.org/10.1126/science.1210214
- Passerini V, Ozeri-Galai E, de Pagter MS, Donnelly N, Schmalbrock S, Kloosterman WP, Kerem B, Storchová Z. The presence of extra chromosomes leads to genomic instability. Nat Commun 2016; 7, 10754; PMID:26876972; http://dx.doi.org/10.1038/ncomms10754
- Ohashi A, Ohori M, Iwai K, Nakayama Y, Nambu T, Morishita D, Kawamoto T, Miyamoto M, Hirayama T, Okaniwa M, et al. Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat Commun 2015; 6, 7668; PMID:26144554; http://dx.doi.org/10.1038/ncomms8668
- Donnelly N, Storchova Z. Aneuploidy and proteotoxic stress in cancer. Mol Cell Oncol 2015; 2(2):e976491; PMID:27308438; http://dx.doi.org/10.4161/23723556.2014.976491
- Nicholson JM, Cimini D. Link between aneuploidy and chromosome instability. Int Rev Cell Mol Biol 2015; 315, 299-317; PMID:25708466
- Fenech M. The in vitro micronucleus technique. Mutat Res 2000; 455(1-2):81-95; PMID:11113469; http://dx.doi.org/10.1016/S0027-5107(00)00065-8
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012; 482(7383):53-8; PMID:22258507; http://dx.doi.org/10.1038/nature10802
- Norden C, Mendoza M, Dobbelaere J, Kotwaliwale CV, Biggins S, Barral Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell 2006; 125(1):85-98; PMID:16615892; http://dx.doi.org/10.1016/j.cell.2006.01.045
- Mendoza M, Norden C, Durrer K, Rauter H, Uhlmann F, Barral Y. A mechanism for chromosome segregation sensing by the NoCut checkpoint. Nat Cell Biol 2009; 11(4):477-83; PMID:19270692; http://dx.doi.org/10.1038/ncb1855
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009; 136(3):473-84; PMID:19203582; http://dx.doi.org/10.1016/j.cell.2008.12.020
- Amaral N, Vendrell A, Funaya C, Idrissi FZ, Maier M, Kumar A, Neurohr G, Colomina N, Torres-Rosell J, Geli MI, et al. The Aurora-B-dependent NoCut checkpoint prevents damage of anaphase bridges after DNA replication stress. Nat Cell Biol 2016; 18(5):516-26; PMID:27111841; http://dx.doi.org/10.1038/ncb3343
- Amaral N, et al. DNA Replication Stress: NoCut to the rescue. Cell Cycle 2017; 16(3):233-234
- Pampalona J, Roscioli E, Silkworth WT, Bowden B, Genescà A, Tusell L, Cimini D. Chromosome Bridges Maintain Kinetochore-Microtubule Attachment throughout Mitosis and Rarely Break during Anaphase. PLoS One 2016; 11(1):e0147420; PMID:26784746; http://dx.doi.org/10.1371/journal.pone.0147420
- Shimizu N, Shingaki K, Kaneko-Sasaguri Y, Hashizume T, Kanda T. When, where and how the bridge breaks: anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp Cell Res 2005; 302(2):233-43; PMID:15561104; http://dx.doi.org/10.1016/j.yexcr.2004.09.001
- Mackay DR, Ullman KS. ATR and a Chk1-Aurora B pathway coordinate postmitotic genome surveillance with cytokinetic abscission. Mol Biol Cell 2015; 26(12):2217-26; PMID:25904336; http://dx.doi.org/10.1091/mbc.E14-11-1563
- Jasencakova Z, Groth A. Replication stress, a source of epigenetic aberrations in cancer? Bioessays 2010; 32(10):847-55; PMID:20726011; http://dx.doi.org/10.1002/bies.201000055
- Jasencakova Z, Groth A. Restoring chromatin after replication: how new and old histone marks come together. Semin Cell Dev Biol 2010; 21(2):231-7; PMID:19815085; http://dx.doi.org/10.1016/j.semcdb.2009.09.018
- Dabin J, Fortuny A, Polo SE. Epigenome Maintenance in Response to DNA Damage. Mol Cell 2016; 62(5):712-27; PMID:27259203; http://dx.doi.org/10.1016/j.molcel.2016.04.006
- Alabert C, Groth A. Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 2012; 13(3):153-67; PMID:22358331; http://dx.doi.org/10.1038/nrm3288
- Khurana S, Oberdoerffer P. Replication Stress: A Lifetime of Epigenetic Change. Genes (Basel) 2015; 6(3):858-77; PMID:26378584
- Sarkies P, Reams C, Simpson LJ, Sale JE. Epigenetic instability due to defective replication of structured DNA. Mol Cell 2010; 40(5):703-13; PMID:21145480; http://dx.doi.org/10.1016/j.molcel.2010.11.009
- Jasencakova Z, Scharf AN, Ask K, Corpet A, Imhof A, Almouzni G, Groth A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol Cell 2010; 37(5):736-43; PMID:20227376; http://dx.doi.org/10.1016/j.molcel.2010.01.033
- Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 2003; 422(6934):909-13; PMID:12712207; http://dx.doi.org/10.1038/nature01596
- Nyce J, Liu L, Jones PA. Variable effects of DNA-synthesis inhibitors upon DNA methylation in mammalian cells. Nucleic Acids Res 1986; 14(10):4353-67; PMID:3086840; http://dx.doi.org/10.1093/nar/14.10.4353
- Tran V, Lim C, Xie J, Chen X, et al. Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 2012; 338(6107):679-82; PMID:23118191; http://dx.doi.org/10.1126/science.1226028
- Gaillard H, García-Muse T, Aguilera A, et al. Replication stress and cancer. Nat Rev Cancer 2015; 15(5):276-89; PMID:25907220; http://dx.doi.org/10.1038/nrc3916
- Burrow AA, Williams LE, Pierce LC, Wang YH, et al. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics 2009; 10, 59; PMID:19183484; http://dx.doi.org/10.1186/1471-2164-10-59
- Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, et al. Signatures of mutation and selection in the cancer genome. Nature 2010; 463(7283):893-8; PMID:20164919; http://dx.doi.org/10.1038/nature08768
- Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, Debatisse M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep 2013; 4(3):420-8; PMID:23911288; http://dx.doi.org/10.1016/j.celrep.2013.07.003
- Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol 2015; 10, 425-48; PMID:25621662; http://dx.doi.org/10.1146/annurev-pathol-012414-040424
- Ma K, Qiu L, Mrasek K, Zhang J, Liehr T, Quintana LG, Li Z. Common fragile sites: genomic hotspots of DNA damage and carcinogenesis. Int J Mol Sci 2012; 13(9):11974-99; PMID:23109895; http://dx.doi.org/10.3390/ijms130911974
- Gao G, Smith DI. Very large common fragile site genes and their potential role in cancer development. Cell Mol Life Sci 2014; 71(23):4601-15; PMID:25300511; http://dx.doi.org/10.1007/s00018-014-1753-6
- Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996; 84(4):587-97; PMID:8598045; http://dx.doi.org/10.1016/S0092-8674(00)81034-X
- Waters CE, Saldivar JC, Hosseini SA, Huebner K. The FHIT gene product: tumor suppressor and genome “caretaker.” Cell Mol Life Sci 2014; 71(23):4577-87; PMID:25283145; http://dx.doi.org/10.1007/s00018-014-1722-0
- Bednarek AK, Keck-Waggoner CL, Daniel RL, Laflin KJ, Bergsagel PL, Kiguchi K, Brenner AJ, Aldaz CM. WWOX, the FRA16D gene, behaves as a suppressor of tumor growth. Cancer Res 2001; 61(22):8068-73; PMID:11719429
- Aqeilan RI, Croce CM. WWOX in biological control and tumorigenesis. J Cell Physiol 2007; 212(2):307-10; PMID:17458891; http://dx.doi.org/10.1002/jcp.21099
- Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, Trapasso F, Drusco A, Shimizu M, Masciullo V, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A 2003; 100(10):5956-61; PMID:12719539; http://dx.doi.org/10.1073/pnas.0931262100
- Gong Y, Zack TI, Morris LG, Lin K, Hukkelhoven E, Raheja R, Tan IL, Turcan S, Veeriah S, Meng S, et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet 2014; 46(6):588-94; PMID:24793136; http://dx.doi.org/10.1038/ng.2981
- Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell 1997; 89(2):215-25; PMID:9108477; http://dx.doi.org/10.1016/S0092-8674(00)80201-9
- Ciullo M, Debily MA, Rozier L, Autiero M, Billault A, Mayau V, El Marhomy S, Guardiola J, Bernheim A, Coullin P, et al. Initiation of the breakage-fusion-bridge mechanism through common fragile site activation in human breast cancer cells: the model of PIP gene duplication from a break at FRA7I. Hum Mol Genet 2002; 11(23):2887-94; PMID:12393800; http://dx.doi.org/10.1093/hmg/11.23.2887
- Hellman A, Zlotorynski E, Scherer SW, Cheung J, Vincent JB, Smith DI, Trakhtenbrot L, Kerem B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 2002; 1(1):89-97; PMID:12086891; http://dx.doi.org/10.1016/S1535-6108(02)00017-X
- Mazouzi A, Velimezi G, Loizou JI. DNA replication stress: causes, resolution and disease. Exp Cell Res 2014; 329(1):85-93; PMID:25281304; http://dx.doi.org/10.1016/j.yexcr.2014.09.030
- Constantinou A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma 2012; 121(1):21-36; PMID:22057367; http://dx.doi.org/10.1007/s00412-011-0349-2
- Porfirio B, Smeets D, Beckers L, Caporossi D, Tedeschi B, Vernole P, Joenje H, Nicoletti B, Dallapiccola B. Fragile sites and chromosome instability: the distribution of breaks induced by cis-diamine-dichloro-platinum (II) in Fanconi anemia lymphocyte cultures. Hum Genet 1991; 86(3):256-60; PMID:1997377; http://dx.doi.org/10.1007/BF00202404
- Fundia A, Gorla N, Larripa I. Spontaneous chromosome aberrations in Fanconi's anemia patients are located at fragile sites and acute myeloid leukemia breakpoints. Hereditas 1994; 120(1):47-50; PMID:8206783; http://dx.doi.org/10.1111/j.1601-5223.1994.00047.x
- Schoder C, Liehr T, Velleuer E, Wilhelm K, Blaurock N, Weise A, Mrasek K. New aspects on chromosomal instability: chromosomal break-points in Fanconi anemia patients co-localize on the molecular level with fragile sites. Int J Oncol 2010; 36(2):307-12; PMID:20043063
- Filipovic J, Joksić G, Vujić D, Joksić I, Mrasek K, Weise A, Liehr T. First molecular-cytogenetic characterization of Fanconi anemia fragile sites in primary lymphocytes of FA-D2 patients in different stages of the disease. Mol Cytogenet 2016; 9(1):70; PMID:27625703; http://dx.doi.org/10.1186/s13039-016-0280-6
- Pawlikowska P, Fouchet P, Vainchenker W, Rosselli F, Naim V. Defective endomitosis during megakaryopoiesis leads to thrombocytopenia in Fanca-/- mice. Blood 2014; 124(24):3613-23; PMID:25261197; http://dx.doi.org/10.1182/blood-2014-01-551457
- Mohaghegh P, Hickson ID. DNA helicase deficiencies associated with cancer predisposition and premature ageing disorders. Hum Mol Genet 2001; 10(7):741-6; PMID:11257107; http://dx.doi.org/10.1093/hmg/10.7.741
- Pirzio LM, Pichierri P, Bignami M, Franchitto A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol 2008; 180(2):305-14; PMID:18209099; http://dx.doi.org/10.1083/jcb.200705126
- Wei PC, Chang AN, Kao J, Du Z, Meyers RM, Alt FW, Schwer B. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016; 164(4):644-55; PMID:26871630; http://dx.doi.org/10.1016/j.cell.2015.12.039
- Yuce O, West SC. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol 2013; 33(2):406-17; PMID:23149945; http://dx.doi.org/10.1128/MCB.01195-12
- Andriani GA, Vijg J, Montagna C. Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech Ageing Dev 2016; in press; PMID:27013377