
ABSTRACT
Acute myeloid leukemias driven by MLL fusion proteins are commonly associated with poor prognosis and refractory treatment. Here, we provide evidence that olaparib can potentiate sensitivity of MLL leukemia cells to conventional chemotherapy and DNMT inhibitors offering new potential therapeutic strategies for MLL rearranged leukemias Using the primary mouse leukemia cells and human MLL-AF9 leukemic cell line, we demonstrate that treatment of MLL-AF9 leukemic cells with DNMT inhibitors or chemotherapies in combination with olaparib results in significant reduction in colony formation or cell growth while the single agent treatments had little impacts. Combining olaparib with DNMT inhibitor induce cell cycle block and apoptosis. Furthermore, we observe a significant increase in proportion of cells with >10 γH2AX DNA damage foci and double stranded breaks when treated with DNMT inhibitors or chemotherapy agents in combination with olaparib, thus providing possible mechanistic insights for the synergism.
Introduction
Leukemias harboring chromosomal 11q23 translocations involving mixed lineage leukemia gene (MLL) account for up to 70% of leukemias in infants lymphoblastic leukemia (ALL) and acute myelogenous leukemia (AML).Citation1 It has been well established that infant ALL with a MLL translocation is associated particularly poor prognosis, with a 4-year event-free survival of approximately 10% as compared to 65% overall survival for patients without MLL translocation.Citation2 Similarly, MLL-rearrangement in adult AML correlates with poor prognosis and inferior treatment response.Citation3 Despite significant increase in our understanding about the biology of the disease, current therapy still revolves around a limited number of intensive chemotherapies, which patients suffer from high frequency of treatment-related deaths and long-term side-effects. Thus, there is clearly a need to develop new and less toxic approaches to treat this disease.
DNMT inhibitors, 5-azacytidine and decitabine were first developed as classic cytostatic agent.Citation4 Since then, it was shown that these compounds inhibit DNA methylation in human cell lines, which provided a mechanistic insight for their differentiation-modulating activity.Citation5 This has led to the use of 5-azacytidine and decitabine as epigenetic drugs which has shown significant clinical benefits in the treatment of myelodysplastic syndrome (MDS).Citation6,7 While, a significant proportion of research focuses on the ability of DNMT inhibitors to reactivate silenced growth-regulatory genes, tumor suppressor genesCitation8 or retroviral elements for triggering immune responses,Citation9,10 a second and less well defined model for DNMT inhibitors' antitumor activity relates to formation of covalent DNMT-DNA adducts, leading to DNA damage and cytotoxicity.Citation11
The Poly ADP ribose polymerase (PARP) family comprises a number of proteins that catalyze the transfer of ADP-ribose to targeted proteins using NAD+ as substrate resulting in mono- or poly(ADP-ribosylation) of targeted proteins.Citation12 Among the PARP family, PARP1 is responsible for most of the total cellular PARylation activity and it controls a wide array of cellular processes, such as DNA repair, gene transcription, cell division and cell death.Citation13 The best well characterized role of PARP1 is its involvement in base excision repair (BER) in the DNA damage response pathway (DDR). PARP1 is able to detect and bind to single strand breaks (SSBs), leading to opening of chromatin and recruit other BER factor to the damage site.Citation14,15 A number of studies have reported synthetic lethality approach of using PARP inhibitors (PARPi) as a potential cancer treatment for ovarian and breast cancers with BRCA1 and BRCA2 mutations.Citation16-22 DNA repair and survival of PARP inhibited cells are heavily dependent of homologous recombination (HR), which are compromised in cancer cells carrying BRCA mutations and thus leading to their susceptibility to PARPi treatment.
More recently, we reported that synthetic lethality induced by PARPi as a possible therapeutic approach in subtypes of AML.Citation23 We found that leukemic cell harboring oncogenic transcription factors AML1-ETO and PML-RARα were sensitive to PARPi treatment both in vitro and in vivo.Citation23 AML1-ETO and PML-RARα suppress the HR transcriptional programs in leukemia cells where PARP inhibition results in accumulation of DNA damage leading to cell differentiation and cell death.Citation23 In stark contrast, leukemia driven by MLL fusion proteins that activate expression of HR genes in part via HOXA9 are resistant to PARPi treatment.Citation23 We showed that modulation of HOXA9 transcription activity could synergize with PARPi in targeting MLL rearranged leukemia. Suppression of Hoxa9 by genetic approach or inhibiting one of its co-regulators, glycogen synthase kinase 3 (GSK3) could sensitize MLL rearranged leukemic cells to PARPi treatment.Citation23 Therefore, the findings provide the rationale and a novel avenue of targeting oncogenic transcriptional factors by PARPi.
In addition to the synthetic lethality approach, the potential use of PARPi, in conjunction with DNA damaging agent have been reported using different model system including AML.Citation24-28 PARPi can enhance the cytotoxic effect of various DNA damaging agent via its inhibition of SSB repair. A number of clinical trials are in progress assessing the safety and efficacy of PARPi with cytotoxic agent in various type of cancers. Here, we set out to explore whether the potential efficacy of PARPi treatment in conjugation with current therapies to target MLL leukemia
Results
Combination of DNMT inhibitors and olaparib
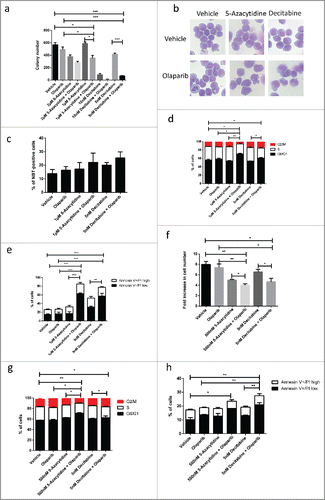
To explore the therapeutic potentials of using PARPi with DNMT inhibitors in MLL leukemia, we tested the colony formation capability of mouse MLL-AF9 primary leukemic cells with two independent DNMT inhibitors, which are commonly used for treatment of MDS and AML.Citation29 We previously identified that in vitro maximal tolerable dose of olaparib is 1µM which exhibited minimal effects on normal primary bone marrow cells.Citation23 MLL-AF9 leukemic cells were treated with 2 different doses of DNMT inhibitors while the dose of the olaparib were kept constant. While olaparib or DNMT inhibitor alone had relative mild impact on the colony number, combination treatments significantly suppressed colony forming capability of the cells especially with lower doses of DNMT inhibitors (), highlighting its potential therapeutic utility. At high concentration, DNMT inhibitors alone could supress colony formation of MLL leukemic cell, and their colony forming capability is further suppressed in the presence olaparib (). We next investigated the cellular processes being affected by combination treatment in MLL-AF9 leukemic cells that might explain the inhibitory effect. Combination of olaparib and DNMT inhibitors did not result in their morphological differentiation as compared to the single agent treatment or untreated control (). This finding was consistent with NBT reduction assay where combination treatment had little impact on the percentage of NBT positive cell (). In contrast, the combination treatment resulted in reduction in cell cycle and induced significant apoptosis (). To investigate if PARPi could exert similar inhibitory effects on the corresponding human leukemias, we used patient derived MLL-AF9 leukemic cell line (MOLM13) for the inhibitor studies. Analogous to the observation in the mouse primary leukemic cells, combination treatment of olaparib and DNMT inhibitors further inhibited cell growth when compared to the single therapy (), resulting in cell cycle arrest () and increase in apoptosis (). Together, these results consistently suggest a potential utility of combining PARPi and DNMT inhibitors for MLL leukemia treatment.
Figure 1. Olaparib potentiates anti-leukaemogenic activity of DNMT inhibitor in MLL leukemia. (a) Quantification of the number of colonies formed by MLL-AF9 LSCs in varying concentration of DNMT inhibitor and/or in combination with 1 µM olaparib. Unpaired t-test was performed between samples. Statistical significances are as indicated, * p < 0.05, *** p < 0.001. (b) Cell morphology of MLL-AF9 LSCs treated with DNMT inhibitor, olaparib or in combination. (c) Data shows percentage of cell that undergoing differentiation characterized by NBT-positive following treatment with DNMT inhibitor, olaparib or in combination. (d) Summary of cell cycle analysis showing relative percentage of cells in G0/G1, S and G2/M phases. Unpaired t-test was performed between samples. Statistical significances are as indicated, * p < 0.05, ** p < 0.01. (e) Quantification of percentage of Annexin V positive cells treated with chemotherapy treatments and/or in combination with olaparib Unpaired t-test was performed between samples. Statistical significances are as indicated, ** p < 0.01, ***p < 0.001. (f) Relative proliferation of patient-derived MLL-AF9 leukemic cell line, MOLM13 treated with DNMT inhibitor, olaparib or in combination Unpaired t-test was performed between samples. Statistical significances are as indicated, *p < 0.05, ** p < 0.01. (g) Summary of cell cycle analysis showing relative percentage of MOLM13 cells in G0/G1, S and G2/M phases after treatment. Unpaired t-test was performed between indicated samples. Statistical significances are as indicated, *p < 0.05, ** p < 0.01. (h) Quantification of percentage of Annexin V positive MOLM13 cells treated DNMT inhibitor, olaparib or in combination. Unpaired t-test was performed between samples. Statistical significances are as indicated, *p < 0.05, ** p < 0.01.
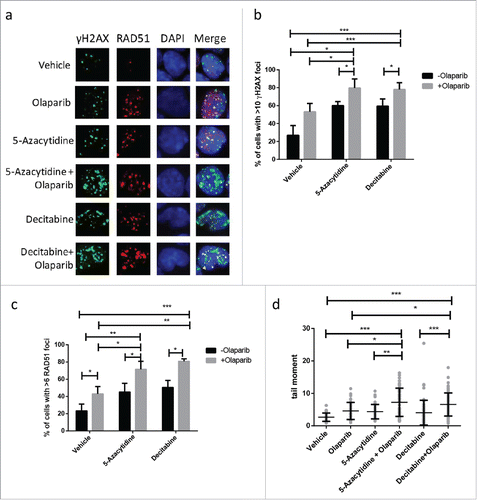
To investigate the effects of the treatments on DNA repair, we assayed DNA damage in the mouse primary leukemic cells by analyzing the frequency of Ser-139 phosphorylated γH2AX foci, which is considered as an early cellular response to DNA damage.Citation30 The percentage of cells with > 10 γH2AX foci was significant higher in the combined treatment while olapariab or DNMT inhibitors had moderate/mild impact on γH2AX foci formation 24h after treatment, suggesting accumulation of DNA damage (). To determine whether DNMT inhibitor could affect HR, the recruitment of RAD51 to DNA damage sites were measured. MLL-AF9 leukemic cell was able to recruit RAD51 protein onto the DNA damage site efficiently upon olaparib treatment at 24 h in the presence or absence of DNMT inhibitors thus suggesting that RAD51 recruitment is not impaired (). However, we did note significantly higher proportion of cells with > 6 RAD51 foci in the combined treatments (). To further investigate the nature of the DNA damage induced by DNMT inhibitors in combination with olaparib, we employed the neutral comet assay to examine the level of double stranded breaks at the individual cellular level. In accordance to γH2AX and RAD51 staining, treatment of mouse MLL-AF9 leukemic cells with DNMT inhibitors in combination with olaparib resulted in significant increase in level of double stranded breaks as measured by comet tail moment ().
Figure 2. Olaparib enhances DNA damage induced with DNMT inhibitor in MLL leukemia. (a) Representative picture of γH2AX and RAD51 immunofluorescent staining of MLL-AF9 LSC treated with DNMT inhibitor, olaparib or in combination for 24 h. (b-c) Quantification of percentage of MLL-AF9 leukemic cells with greater than 10 γH2AX and greater than 6 RAD51 foci following treatment as indicated. Unpaired t-test was performed between indicated samples. Statistical significances are as indicated, *p < 0.05, ** p < 0.01, ***p < 0.001. (d) Quantification of comet tail formation in MLL-AF9 LSC following 24 h treatment of DNMT inhibitor, olaparib or in combination. Results are presented tail moment. At least 50 cells were analyzed for each condition. Unpaired t-test was performed between indicated samples. Statistical significances are as indicated, *p < 0.05, ** p < 0.01, ***p < 0.001.
Combining olaparib with chemotherapy
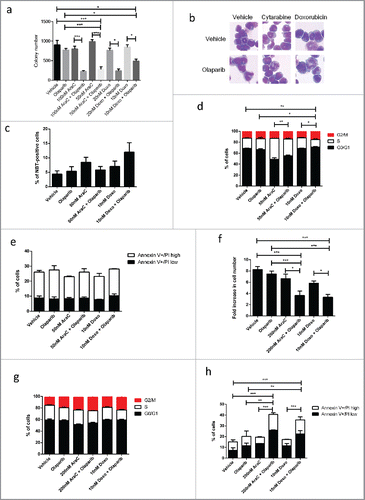
To examine whether PARPi could sensitize MLL leukemic cell to standard chemotherapy, we also tested the colony forming capability of MLL-AF9 leukemic cells with two independent chemotherapy agents commonly used in AML, doxorubicin and cytarabine (AraC). Similarly, to DNMT inhibitors, we opted for two differently doses of the chemotherapy agents and kept concentration of olaparib constant. Olaparib and chemotherapy agent alone had relative mild or no impact on colony number, however, combining chemotherapy with olaparib significantly impaired colony forming ability (). Combination therapy did not result in their morphological differentiation or increase in NBT positive staining (). Interestingly, combination between doxorubicin and olaparib significantly impaired cell cycle whereas cytarabine with olaparib did not exhibit the same effect (). This might be potentially related to their different mechanisms of action whereby doxorubicin acts predominantly by inhibiting DNA-associated enzymes such topoisomerase enzymes and intercalating the base pairs of the DNA's double helix,Citation31 whereas cytarabine exerts its cytotoxicity by inhibiting DNA polymerase or by terminating chain elongation upon incorporation into newly synthesized DNA.Citation32,33 Similarly, combination of PARPi with chemotherapy could also exert significant inhibitory effects on the corresponding human leukemias cell line, by inhibiting cell number () and induce apoptosis while having little impact on the cell cycle ().
Figure 3. Olaparib potentiates anti-leukaemogenic activity of conventional chemotherapy in MLL leukemia. (a) Quantification of the number of colonies formed by MLL-AF9 LSCs in varying concentration of chemotherapy agents and/or in combination with 1 µM olaparib. Unpaired t-test was performed between indicated samples. Statistical significances are as indicated, *p < 0.05, ***p < 0.001. (b) Cell morphology of MLL-AF9 LSCs treated with chemotherapy agents, olaparib or in combination. (c) Data shows percentage of cell that undergoing differentiation characterized by NBT-positive following treatment with chemotherapy agents, olaparib or in combination. (d) Summary of cell cycle analysis showing relative percentage of cells in G0/G1, S and G2/M phases are indicted. Unpaired t-test was performed between samples, Statistical significances are as indicated, *p < 0.05, ** p < 0.01. (e) Quantification of percentage of Annexin V positive cells treated with chemotherapy treatments and/or in combination with olaparib. (f) Relative proliferation of patient-derived MLL-AF9 leukemic cell line, MOLM13 treated with chemotherapy, olaparib or in combination. Unpaired t-test was performed between indicated samples. Statistical significances are as indicated, ** p < 0.01, ***p < 0.001. (g) Summary of cell cycle analysis showing relative percentage of MOLM13 cells in G0/G1, S and G2/M phases after treatment. (h) Quantification of percentage of Annexin V positive MOLM13 cells treated with chemotherapy, olaparib or in combination. Unpaired t-test was performed between samples. Statistical significances are as indicated ** p < 0.01, ***p < 0.001.
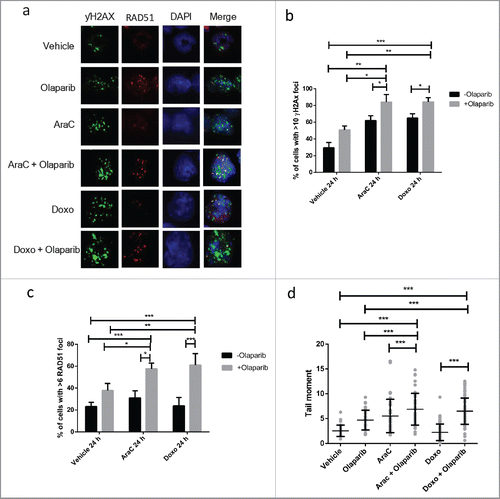
To explore their potential impact on DNA damage response, we again analyzed the formation of γH2AX and RAD51 foci in MLL-AF9 leukemic cells. Combining chemotherapy with olaparib resulted in significant increase in proportion of cell with DNA damage as compared to single agents (). Chemotherapy had no impact on the recruitment of RAD51 to damage foci upon PARP inhibition (). Nevertheless, further analysis of comet tails revealed significant increase in double stranded breaks in the combination treatments as compared to single agents or untreated control ().
Figure 4. Olaparib enhances DNA damage induced with conventional chemotherapy in MLL leukemia. (a) Representative picture of γH2AX and RAD51 immunofluorescent staining of MLL-AF9 LSC treated with chemotherapy, olaparib or in combination for 24 h. (b-c) Quantification of percentage of MLL-AF9 leukemic cells with greater than 10 γH2AX and greater than 6 RAD51 foci upon treatment. Unpaired t-test was performed between samples. Statistical significances are as indicated *p < 0.05, ** p < 0.01, ***p < 0.001. (d) Quantification of comet tail formation in MLL-AF9 LSC following 24 h treatment of chemotherapy, olaparib or in combination. Results are presented tail moment. At least 50 cells were analyzed for each condition. Unpaired t-test was performed between indicated samples. Statistical significances are as indicated, ***p < 0.001.
Discussion
We performed in vitro assays exploring potential use of olaparib in combination with current AML therapy agents and demonstrated that olaparib can sensitize MLL leukemic cells to both DNMT inhibitors and chemotherapy agents. This increases the efficacy of these cytotoxic agents and allows substantial reduction of their dose and consequently their side effects. We also provided mechanistic insight that PARPi in combination with cytotoxic agent results in accumulation double stranded breaks leading to cell cycle arrest or apoptosis and consequently suppression in clongenic potential of MLL leukemia cells.
Extensive studies have shown that PARPi could enhance various cytotoxic agents such as platinum salts,Citation34 temozolomide,Citation35-37 and topoisomerase inhibitorsCitation38 and the mechanism of which is by inhibiting the PARP-mediated repair of these DNA breaks induced by these agents. Additionally, PARP1 knockout also drastic impairs DNA repair following damage caused by ionising radiation or cytotoxic treatment.Citation39 In AML, the combination of PARPi, Rucaparib, and conventional chemotherapy agent, fluroouracil, was effective in killing AML cells in vitro and in vivo as a result of fluroouracil induced DNA damage together with repression of the DDR by PARPi.Citation24 Similarly, PARPi was able sensitize a panel of AML cell lines to 5-azacytidine.Citation25
There are two different non-exclusive cytotoxic mechanisms, which DNMT inhibitors may function; epigenetic changes in gene expression causing cell death and trapping of DNMT onto DNA causing lethal DNA damage and DNA damage generated by randomly incorporated DNMT inhibitors. While the ability of DNMT inhibitors to inhibit cellular DNMTs and mediate re-expression of aberrantly hypermethylated growth-regulatory genes is well known,Citation40 the role of 5-azacytidine and decitabine inducing DNA damage and covalently trapping DNMTs onto DNA is less well defined. Study by Ortal et al. suggests that the incorporation of 5-azacytidine into the DNA is repaired via the BER pathway in which PARP plays a role.Citation25 PARP inhibited cells or BER deficient cells were sensitive to 5-azacytidine treatment leading to increase in γH2AX. Additionally, 5-azacytidine has been proposed to directly induce double stranded breaks by covalently trapping DNMT onto the DNA, which results in replication fork collapse and formation of one-ended DSBs.Citation41,42 Consistently, exposure of multiple myleoma cells to 5-azacytidine results in activation of ATM/ATR HR pathways and their downstream effector responses, such as CHK1 and CHK2.Citation43
The mechanism of action for chemotherapy agents to induce DNA damage is not fully understood. It is likely that they induced DNA damage via multiple mechanisms. For example, doxorubicin is predominantly considered as a classical topoisomerase II inhibitor.Citation31 The topoisomerase family of enzymes catalyze the unwinding of DNA during replication which involves cleavage of one strand of the DNA duplex and passing a second duplex through this transient cleavage. Doxorubicin inhibits topoisomerase II upon cleavage and prevents religation of the cleaved duplex that results in a DNA double-strand break.Citation44 However, doxorubicin exhibits a wide spectrum of DNA damage independent topoisomerase IICitation45 including inhibition of DNA and RNA synthesis,Citation46,47 production of free radicals,Citation48 and the formation of formaldehyde-mediated doxorubicin-DNA adducts.Citation49 Thus while we here present that low doses of DNMT inhibitors or chemotherapy agents that induce multiple types of DNA damage can synergize with PARPi leading to an increase double strand break burden of HR pathway, future in vivo studies are likely to be required in order to examine whether combination therapy could be applied in the clinics.
Materials and methods
Culture of cell lines
MOLM13 cell lines were cultured in RPMI (Invitrogen) supplemented with 10% FBS, 2 mM L-Glutamine. Primary mouse leukemic cells were cultured in RPMI (Invitrogen) supplemented with 20% selected FBS, 2 mM L-Glutamine, 20 ng/ml SCF, 10 ng/ml IL-3, 10 ng/ml IL-6.
Drug treatment
Drug studies of mouse cells were carried out by plating 3×103 cells in 1% methylcellulose medium containing 20 ng/ml SCF, 10 ng/ml IL-3, 10 ng/ml IL-6 and 10 ng/ml GM–CSF in the presence or absence of olaparib (LC Laboratories), 5azacytidine (Sigma) or decitabine (Sigma) at the concentrations indicated. Colonies number were acquired 6-7 days after plating.
Cell cycle analysis
For each assay 1×105 cells were collected, washed in PBS and fixed in 70% cold ethanol. After re-hydration with PBS and centrifugation at 500g for five minutes, the cells were incubated with a solution of PBS containing 1% FCS, 40µg/ml RNAse and 500µg/ml propidium iodide solution (Sigma-Aldrich) in the dark for 30 minutes at 37°C. Samples were acquired at the FACS LSRII (BD Biosciences PharMingen) and analyzed with Flowjo.
Annexin V staining
For each assay 105 cells were collected, washed in PBS and re-suspended in Annexin V binding solution (Biolegend). After centrifugation at 500g for five minutes, the cells were incubated with the Annexin V Binding solution containing 0.25 ug/ml mouse Annexin V-APC (Biolegend) and 1µg/ml propidium iodide in the dark for 30 minutes at 4°C. Samples were then resuspended in binding solution analyzed at the FACS LSRII (BD Biosciences PharMingen). Data were analyzed with Flowjo.
May–grunwald-giemsa staining
105 cells were cytospun for 5 min at 300g onto glass slides. Slides were then stained with May–Grunwald solution (Sigma-Aldrich) for 5 min at room temperature. After washing, they were incubated for 20 min in Giemsa solution (Sigma-Aldrich). Slides were washed again in water and air-dried.
Nitro blue tetrazolium (NBT) reduction assay
NBT reduction assay were performed to determine myeloid differentiation. 0.1% of NBT (final concentration) was added to the liquid culture or semi-solid methocult and incubated at 37°C CO2 incubator for 3hrs. Cells were then washed in PBS and the differentiated cells were indicated by the deposition of dark blue insoluble formazan (NBT positive cells) and the percentage of differentiated cells were counted under microscopy. At least 200 cells were counted in most of the cases.
Immunofluorescence staining of yH2AX and RAD51
Cells were spun onto a glass slide at 400g for 5 minutes and then fixed for 30 minutes in 4% PFA and permeabilized and blocked in PBS containing 0.8% Tx-100/10% FBS/1% BSA (Sigma-Aldrich) for 15 min at room temperature. Anti-mouse yH2AX (ser139) (Upstate clone JBW301 #05-636) and anti-mouse Rad51 (Santa Cruz Biotecnology H92 #sc-8349) were diluted in TBS containing 10%FBS/1%BSA and incubated overnight at 4oC. Slides were then washed three times with PBS and subsequently incubated with 1:200 anti-mouse DL 488 (Jackson/Stratech 715-485-150) and 1:200 anti-rabbit Cy3 (Jackson/Stratech 111-165-144) in PBS containing DAPI 0.2 ug/ml, 10%FBS, 1%BSA for 1 hour at room temperature in the dark. Slides were then washed five times at 10 min each with PBS. Slides were briefly rinsed in water and air-dried prior to mounting.
Comet assay
Comet assay was performed using CometAssay® kit (Trevigen) according to the manufacture protocol. For each slide, 50 randomly chosen comets were acquired using fluorescent microscope (Leica) and analyzed using Comet Assay Software Project Lab (CaspLab).
Statistical analysis
Statistical tests were performed on experiments that had been repeated at least 3 times, which is sufficient the effect observed. All the experiments were analyzed using Student's t-test. p-values lower than 0.05 were considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Biondi A, Cimino G, Pieters R, Pui CH. Biological and therapeutic aspects of infant leukemia. Blood 2000; 96:24-33; PMID:10891426
- Behm FG, Raimondi SC, Frestedt JL, Liu Q, Crist WM, Downing JR, Rivera GK, Kersey JH, Pui CH. Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukemia, regardless of presenting age. Blood 1996; 87:2870-7; PMID:8639906
- Zeisig BB, Kulasekararaj AG, Mufti GJ, So CW. SnapShot: Acute myeloid leukemia. Cancer Cell 2012; 22:698-8 e691; http://dx.doi.org/10.1016/j.ccr.2012.10.017
- Sorm F, Piskala A, Cihak A, Vesely J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia 1964; 20:202-3; PMID:5322617; http://dx.doi.org/10.1007/BF02135399
- Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980; 20:85-93; PMID:6156004; http://dx.doi.org/10.1016/0092-8674(80)90237-8
- Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, Stone RM, Nelson D, Powell BL, DeCastro CM, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 2002; 20:2429-40; PMID:12011120; http://dx.doi.org/10.1200/JCO.2002.04.117
- Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006; 106:1794-803; PMID:16532500; http://dx.doi.org/10.1002/cncr.21792
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349:2042-54; PMID:14627790; http://dx.doi.org/10.1056/NEJMra023075
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015; 162:974-86; PMID:26317466; http://dx.doi.org/10.1016/j.cell.2015.07.011
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015; 162:961-73; PMID:26317465; http://dx.doi.org/10.1016/j.cell.2015.07.056
- Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A 1994; 91:11797-801; PMID:7527544; http://dx.doi.org/10.1073/pnas.91.25.11797
- Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci 2010; 35:208-19; PMID:20106667; http://dx.doi.org/10.1016/j.tibs.2009.12.003
- Sousa FG, Matuo R, Soares DG, Escargueil AE, Henriques JA, Larsen AK, Saffi J. PARPs and the DNA damage response. Carcinogenesis 2012; 33:1433-40; PMID:22431722; http://dx.doi.org/10.1093/carcin/bgs132
- Wang Z, Wang F, Tang T, Guo C. The role of PARP1 in the DNA damage response and its application in tumor therapy. Front Med 2012; 6:156-64; PMID:22660976; http://dx.doi.org/10.1007/s11684-012-0197-3
- Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 2010; 39:8-24; PMID:20603072; http://dx.doi.org/10.1016/j.molcel.2010.06.017
- Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 2012; 12:68-78; http://dx.doi.org/10.1038/nrc3181
- Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, Kowalczykowski S, et al. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 2009; 136:1032-43; PMID:19303847; http://dx.doi.org/10.1016/j.cell.2009.02.019
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361:123-34; PMID:19553641; http://dx.doi.org/10.1056/NEJMoa0900212
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010; 376:235-44; PMID:20609467; http://dx.doi.org/10.1016/S0140-6736(10)60892-6
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913-7; PMID:15829966; http://dx.doi.org/10.1038/nature03443
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917-21; PMID:15829967; http://dx.doi.org/10.1038/nature03445
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8:193-204; PMID:18256616; http://dx.doi.org/10.1038/nrc2342
- Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, Gil J, Leung AY, Ashworth A, So CW, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med 2015; 21:1481-90; PMID:26594843; http://dx.doi.org/10.1038/nm.3993
- Falzacappa MV, Ronchini C, Faretta M, Iacobucci I, Di Rorà AG, Martinelli G, Meyer LH, Debatin KM, Orecchioni S, Bertolini F, et al. The Combination of the PARP Inhibitor Rucaparib and 5FU Is an Effective Strategy for Treating Acute Leukemias. Mol Cancer Ther 2015; 14:889-98; PMID:25667168; http://dx.doi.org/10.1158/1535-7163.MCT-14-0276
- Orta ML, Höglund A, Calderón-Montaño JM, Domínguez I, Burgos-Morón E, Visnes T, Pastor N, Ström C, López-lázaro M, Helleday T, et al. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2′-deoxycytidine lesions. Nucleic Acids Res 2014; 42:9108-20; PMID:25074383; http://dx.doi.org/10.1093/nar/gku638
- Faraoni I, Compagnone M, Lavorgna S, Angelini DF, Cencioni MT, Piras E, Panetta P, Ottone T, Dolci S, Venditti A, et al. BRCA1, PARP1 and gammaH2AX in acute myeloid leukemia: Role as biomarkers of response to the PARP inhibitor olaparib. Biochim Biophys Acta 2015; 1852:462-72; http://dx.doi.org/10.1016/j.bbadis.2014.12.001
- Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010; 10:293-301; PMID:20200537; http://dx.doi.org/10.1038/nrc2812
- Zhao L, So CW. PARP-inhibitor-induced synthetic lethality for acute myeloid leukemia treatment. Experimental hematology 2016; 44:902-7; PMID:27473567; http://dx.doi.org/10.1016/j.exphem.2016.07.007
- Lubbert M, Bertz H, Rüter B, Marks R, Claus R, Wäsch R, Finke J. Non-intensive treatment with low-dose 5-aza-2′-deoxycytidine (DAC) prior to allogeneic blood SCT of older MDS/AML patients. Bone Marrow Transplant 2009; 44:585-8; PMID:19363531; http://dx.doi.org/10.1038/bmt.2009.64
- Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 2010; 24:679-86; PMID:20130602; http://dx.doi.org/10.1038/leu.2010.6
- Hilmer SN, Cogger VC, Muller M, Le Couteur DG. The hepatic pharmacokinetics of doxorubicin and liposomal doxorubicin. Drug Metab Dispos 2004; 32:794-9; PMID:15258103; http://dx.doi.org/10.1124/dmd.32.8.794
- Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol 2004; 53:209-19; PMID:14610616; http://dx.doi.org/10.1007/s00280-003-0726-5
- Sampath D, Rao VA, Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene 2003; 22:9063-74; PMID:14663485; http://dx.doi.org/10.1038/sj.onc.1207229
- Michels J, Vitale I, Senovilla L, Enot DP, Garcia P, Lissa D, Olaussen KA, Brenner C, Soria JC, Castedo M, et al. Synergistic interaction between cisplatin and PARP inhibitors in non-small cell lung cancer. Cell Cycle 2013; 12:877-83; PMID:23428903; http://dx.doi.org/10.4161/cc.24034
- Gaymes TJ, Mohamedali AM, Patterson M, Matto N, Smith A, Kulasekararaj A, Chelliah R, Curtin N, Farzaneh F, Shall S, et al. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica 2013; 98:1397-406; PMID:23349304; http://dx.doi.org/10.3324/haematol.2012.079251
- Javle M, Curtin NJ. The potential for poly (ADP-ribose) polymerase inhibitors in cancer therapy. Ther Adv Med Oncol 2011; 3:257-67; PMID:22084640; http://dx.doi.org/10.1177/1758834011417039
- Horton TM, Jenkins G, Pati D, Zhang L, Dolan ME, Ribes-Zamora A, Bertuch AA, Blaney SM, Delaney SL, Hegde M, et al. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Mol Cancer Ther 2009; 8:2232-42; PMID:19671751; http://dx.doi.org/10.1158/1535-7163.MCT-09-0142
- Znojek P, Willmore E, Curtin NJ. Preferential potentiation of topoisomerase I poison cytotoxicity by PARP inhibition in S phase. Br J Cancer 2014; 111:1319-26; PMID:25003660; http://dx.doi.org/10.1038/bjc.2014.378
- Tomoda T, Kurashige T, Moriki T, Yamamoto H, Fujimoto S, Taniguchi T. Enhanced expression of poly(ADP-ribose) synthetase gene in malignant lymphoma. Am J Hematol 1991; 37:223-27; PMID:1907096; http://dx.doi.org/10.1002/ajh.2830370402
- Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 2002; 21:5483-95; PMID:12154409; http://dx.doi.org/10.1038/sj.onc.1205699
- Michel B, Flores MJ, Viguera E, Grompone G, Seigneur M, Bidnenko V. Rescue of arrested replication forks by homologous recombination. Proc Natl Acad Sci U S A 2001; 98:8181-8; PMID:11459951; http://dx.doi.org/10.1073/pnas.111008798
- McGlynn P, Lloyd RG. Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol 2002; 3:859-70; PMID:12415303; http://dx.doi.org/10.1038/nrm951
- Kiziltepe T, Hideshima T, Catley L, Raje N, Yasui H, Shiraishi N, Okawa Y, Ikeda H, Vallet S, Pozzi S, et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol Cancer Ther 2007; 6:1718-27; PMID:17575103; http://dx.doi.org/10.1158/1535-7163.MCT-07-0010
- Liu LF, Rowe TC, Yang L, Tewey KM, Chen GL. Cleavage of DNA by mammalian DNA topoisomerase II. J Biol Chem 1983; 258:15365-70; PMID:6317692
- Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999; 57:727-41; PMID:10075079; http://dx.doi.org/10.1016/S0006-2952(98)00307-4
- Di Marco A, Silvestrini R, Di Marco S, Dasdia T. Inhibiting effect of the new cytotoxic antibiotic daunomycin on nucleic acids and mitotic activity of HeLa cells. J Cell Biol 1965; 27:545-50; PMID:5885429; http://dx.doi.org/10.1083/jcb.27.3.545
- Munger C, Ellis A, Woods K, Randolph J, Yanovich S, Gewirtz D. Evidence for inhibition of growth related to compromised DNA synthesis in the interaction of daunorubicin with H-35 rat hepatoma. Cancer Res 1988; 48:2404-11; PMID:3356005
- Feinstein E, Canaani E, Weiner LM. Dependence of nucleic acid degradation on in situ free-radical production by adriamycin. Biochemistry 1993; 32:13156-161; PMID:7694653; http://dx.doi.org/10.1021/bi00211a026
- Cutts SM, Nudelman A, Rephaeli A, Phillips DR. The power and potential of doxorubicin-DNA adducts. IUBMB Life 2005; 57:73-81; PMID:16036566; http://dx.doi.org/10.1080/15216540500079093