
ABSTRACT
Bcl-2 inhibits cell proliferation by delaying G0/G1 to S phase entry. We tested the hypothesis that Bcl-2 regulates S phase entry through mitochondrial pathways. Existing evidence indicates mitochondrial adenosine tri-phosphate (ATP) and reactive oxygen species (ROS) are important signals in cell survival and cell death, however, the molecular details of how these 2 processes are linked remain unknown. In this study, 2 cell lines stably expressing Bcl-2, 3T3Bcl-2 and C3HBcl-2, and vector-alone PB controls were arrested in G0/G1 phase by serum starvation and contact inhibition, and ATP and ROS were measured during re-stimulation of cell cycle entry. Both ATP and ROS levels were decreased in G0/G1 arrested cells compared with normal growing cells. In addition, ROS levels were significant lower in synchronized Bcl-2 cells than those in PB controls. After re-stimulation, ATP levels increased with time, reaching peak value 1–3 hours ahead of S phase entry for both Bcl-2 cells and PB controls. Consistent with 2 hours of S phase delay, Bcl-2 cells reached ATP peaks 2 hours later than PB control, which suggests a rise in ATP levels is required for S phase entry. To examine the role of ATP and ROS in cell cycle regulation, ATP and ROS level were changed. We observed that elevation of ATP accelerated cell cycle progression in both PB and Bcl-2 cells, and decrease of ATP and ROS to the level equivalent to Bcl-2 cells delayed S phase entry in PB cells. Our results support the hypothesis that Bcl-2 protein regulates mitochondrial metabolism to produce less ATP and ROS, which contributes to S phase entry delay in Bcl-2 cells. These findings reveal a novel mechanistic basis for understanding the link between mitochondrial metabolism and tumor-suppressive function of Bcl-2.
KEYWORDS:
Introduction
Bcl-2 was first found to be a key apoptosis regulator highly expressed in B cell lymphoma in 1985.Citation1 Since then, approximately 25 Bcl-2 gene family members have been identified, composed of anti-apoptotic proteins, including Bcl-2, Bcl-XL, Bcl-w, Mcl-1, A1, Nr-13, etc, and pro-apoptotic proteins, such as Bax, Bak, Bad, Bid, Bim, Noxa, Hrk, and others. Bcl-2 family members share a carboxyl trans-membrane (TM) region and 1∼4 Bcl-2 homologous structure domain (BH1∼4). A large body of studies supports Bcl-2 proteins as the critical arbiters of apoptotic cell death, and the interactions among these proteins regulate the balance between cell death and survival through mitochondrial pathway.Citation2 Dimerization of the proapoptotic executioner proteins BAX and BAK in the mitochondrial outer membrane leads to mitochondrial outer membrane permeabilization, and subsequent release of cytochrome c and additional apoptotic factors, promoting caspase activation, which ultimately culminates in apoptotic cell death.Citation3
Over-expression of Bcl-2 observed in many cancers confirmed Bcl-2 as an oncogene.Citation4 Bcl-2 also functions as a tumor suppressor gene. In mouse and human breast cancer models, Bcl-2 overexpression inhibits glandular cell proliferation, reduces the incidence of tumor, and delays the age of tumor onset.Citation5–7 The dual function of the Bcl-2 protein was observed in Bcl-2 transgenic mouse, in that lymphoid T cell proliferation in young mice were restrained, while the tumor morbidity was increased.Citation7 Therefore, acceleration of tumor cell apoptosis and inhibition of tumor cell proliferation are the important strategies in anti-tumor therapy. As a target gene in cancer therapy, Bcl-2 was intensively studied in recent decades.Citation8,9 However, the precise mechanism by which Bcl-2 exerts tumor suppressor function is not fully understood.
The recent interest in aerobic glycolysis in cancer cells raises possibilities that metabolic states in cancer cells might be associated with Bcl-2's anti-tumor function. ROS plays an important role in cell cycle in plants and mammals,Citation10,11 and low ROS was found to inhibit the cellular growth and proliferation.Citation12,13 Bcl-2 overexpression could inhibit cell proliferation through delay of G0/G1 to S phase process via the mitochondrial pathway.Citation14–16 ATP and ROS are produced in mitochondrial oxidative phosphorylation to maintain homeostasis, and change of ATP and ROS levels could affect cell proliferation and mediate tumor development.Citation17,18
Bcl-2 was initially reported to have anti-oxidant effects in 1993. Bcl-2-expressing cells are intrinsically resistant to added oxidative stress.Citation19,20 Other studies suggested that ROS, in particular hydrogen peroxide (H2O2), decrease the expression of Bcl-2 and increase the expression of pro-apoptotic proteins to modulate apoptosis.Citation21,22 Our preliminary study found that ATP and ROS level were regulated by Bcl-2 protein in cell cycle, leading to the hypothesis that ATP and ROS might play key roles in cell cycle regulation by Bcl-2. In this study, we focused on the levels of ATP and ROS from G0/G1 to S phase entry, and their relationship with Bcl-2s cell cycle function.
Results
S phase entry delay in Bcl-2 stable expressing cells than PBabe control
PBabe (PB) and Bcl-2 virus were produced from 293T cells, and used to infect 3T3 or C3H cells. 3T3Bcl-2 and C3HBcl-2 with their PB vector control cell lines were successfully established ().
Figure 1. Cell cycle profiles and p27 expression in synchronized 3T3PB and Bcl−2 cells by SS or CI followed by re-stimulation. (A) Bcl−2 expression in 293T, 3T3 and C3H cells. (B, C) Cell cycle profile of 3T3PB and Bcl−2 cells (D, E) Percentage of S phase cells during re-stimulation in serum-starved SS3T3 and contact inhibited 3T3 cells. (F, G). Western blot of p27 in normal growing and synchronized cells. PB: PBABE empty vector stable transfected; Bcl−2: pBABEneo-Bcl−2 stable transfected. CI: contact inhibition SS: serum starvation NG: normal growing
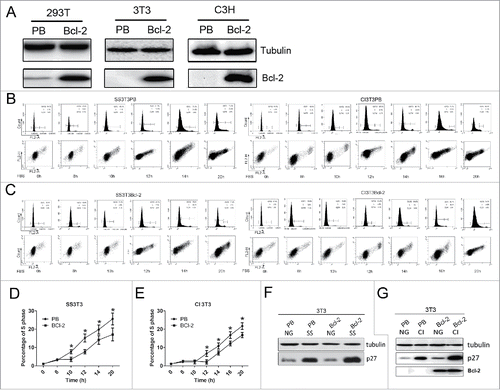
The tumor repressive function of Bcl-2 is mainly the delay of S phase entry from G0/G1.Citation14 To validate this function in our 3T3Bcl-2 and C3HBcl-2 cell lines, contact inhibition (CI) and serum starvation (SS) methods were performed to synchronize cells. Cell cycle profiling showed that more than 90% of cells were arrested in G0/G1 phase under CI or SS treatment (0 hr in ). During re-stimulation, synchronized Bcl-2 cells delayed S phase entry by 2 hrs compared with PB controls (8–20hr in ). Western blot demonstrated that p27 expression was higher in Bcl-2 cells than in PB controls (), consistent with our previous report that Bcl-2 delay cell cycle progression by stabilization of p27 in arrested cells.Citation16 Taken together, the results in confirmed that Bcl-2 delayed cell cycle entry in these cell lines. Next, we used these cell lines to investigate the underlying mechanisms mediating Bcl-2s cell cycle function.
ATP is required for S phase entry
We measured ATP levels at different time points during re-stimulation and observed that ATP increased with time for both 3T3Bcl-2 and PBabe control cells. ATP content in PB cells reached peak value at 8hr and cells entered into S phase at 9hr. ATP content in Bcl-2 cells reached peak value at 12hr and cells entered into S phase at 14hr. The ATP peak was lower in Bcl-2 cells than that in PBabe control cells (). In conclusion, ATP levels reached peak values 1–2hrs before S phase entry for both cell lines, and ATP peak was lower and S phase entry was correspondingly delayed for Bcl-2 cells. These results strongly indicated that ATP content was related to S phase entry, and S phase entry might require a certain level of ATP.
Figure 2. The effects of ATP alteration on cell cycle arrest in 3T3PB and 3T3Bcl−2 cells. (A)The ATP levels during re-stimulation. (B) Cell cycle profiles with addition of various dose of ATP sodium salt after re-stimulation. (C) Graph of the percentage of S phase cells in B. (D) Cell cycle profiles with oligomycinA treatment after re-stimulation. (E) Graph of the percentage of S phase cells in B. (F) Detection of ATP level with oligomycinA treatment.
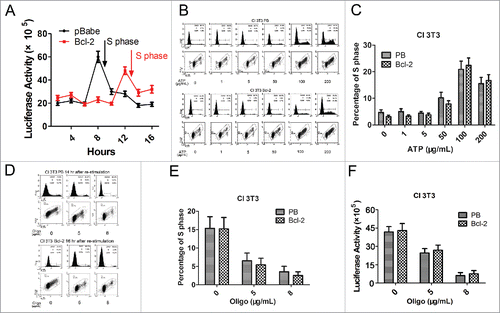
To accelerate S phase entry, we add exogenous ATP sodium salt 1–200 μg/ml in arrested PB and Bcl-2 cells respectively. The results showed that elevating ATP level with 100 or 200 μg/ml of ATP sodium salt promoted S phase comparing with 0–50μg/ml treatment, indicating that S phase entry demands a certain amount of ATP (). ATP is generated from Electron Transport Chain (ETC), composed of complexes in mitochondrial inner membrane. To demonstrate that reduced ATP level impedes cell cycle progression, oligomycin A was added as complex V inhibitor to block ATP production in both PB and Bcl-2 cells during re-stimulation. We found that arrested PB cells enter S phase (∼15%) at 14hr after re-stimulation without oligomycin A treatment, 5 and 8μg/ml oligomycin A treatment block arrested PB cells to enter S phase (less than 5%). Consistently, 5 and 8μg/ml oligomycin A treatment inhibit arrested Bcl-2 cells to enter S phase at 16 hr after re-stimulation, when Bcl-2 cells enter S phase without oligomycin A treatment (). In these treatments, we detected less 5% of cell death in both PB and Bcl-2 cells, which exclude the possible toxic effects of oligomycin A on these cells. proved that 5 and 8μg/ml oligomycin A treatment decreased the ATP level, which in turn leads to S phase entry delay. Thus, these results supported that a rise in ATP is essential to S phase entry.
ROS is a key signal to promote S phase entry
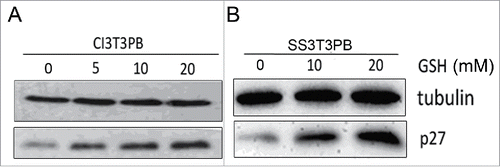
We further asked if mitochondrial ROS was involved in Bcl-2s cell cycle function. Mitochondrial ROS specific fluorescent dye mitoSOX, was chosen to stain the CI or SS 3T3 cells. Flow cytometry analysis read lower ROS signal for Bcl-2 cells compared with PB controls (), which suggests less ROS in Bcl-2 cells might delay cell growth. So we conducted later experiments to reduce ROS level in arrested PB cells to mimic Bcl-2 cells, then to identify whether less ROS delay S phase entry as that occurred in Bcl-2 cells. We first use GSH (ROS scavenger) to diminish ROS contents during re-stimulation in PB cells, flow cytometry analysis confirmed that supplementation of 20 mmol/L GSH reduce ROS equivalent to Bcl-2 cells ().
Figure 3. Detection of ROS levels by flow cytometer. (A) ROS levels in synchronized 3T3PB and Bcl−2 cells by CI treatment. (B) ROS levels in synchronized 3T3PB and Bcl−2 cells by SS treatment. (C) Reduction of ROS level by 20 mmol/L GSH treatment.
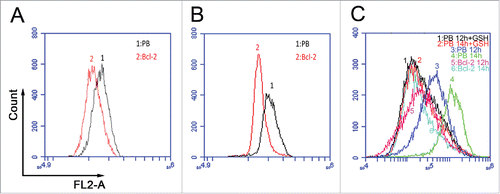
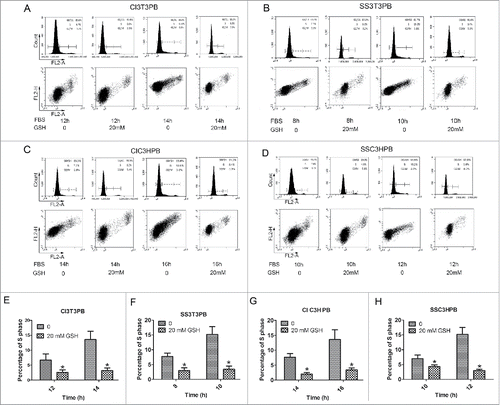
Next, we found that addition of 20 mmol/L GSH into arrested PB cells resulted in S phase delay () during re-stimulation. Decreasing ROS mimicked the ROS scavenger function of Bcl-2, delaying cell cycle progression as in Bcl-2 cells. This finding is consistent with Bcl-2s cell cycle delay function being mediated by ROS. We failed to accelerate S phase entry by adding H2O2 to Bcl-2 cells (data not shown), which also suggests that Bcl-2 could clear excessive ROS.
Figure 4. The (S)phase entry profile for PB cells w/o GSH addition during re-stimulation. (A, B) Cell cycle profiles of 3T3PB cells by CI or SS. (C, D) Cell cycle profile of C3HPB cells by CI or SS. (E–F) Graph of percentage of S phase cells in A-D.
P27 was upregulated by decreasing ROS
P27 stabilization is the main mediator of Bcl-2s cell cycle functionCitation16 We checked p27 expression in GSH treated cells to explore the signaling pathway of ROS. CI and SS treated PB cells contained more ROS and less p27 than Bcl-2 cells. When we decreased ROS level by adding GSH to mimic Bcl-2 cells, western blotting showed that p27 was upregulated by in a dose dependent manner (). These results suggest a role of ROS in pP27 expression, which in turn regulates cell cycle progression.
Figure 5. P27 was upregulated by adding 0, 5, 10, 20 mM of GSH.
Discussion
The pro-apoptotic and anti-apoptotic members of the Bcl-2 family are critical regulators in cancer pathogenesis and clinical outcome.Citation23,24 Bcl-2 promotes cell survival by interfering with Bax/Bak proteins through the mitochondrial pathway. Targeting mitochondrial metabolism has recently emerged as a promising approach in cancer treatment, as the energy generation and ROS signaling are involved in survival and growth of cancer cells.
In this study, we focused on the relation between mitochondrial metabolism and Bcl-2s anti-cancer function, which is mediated by delay of cell cycle progression. Our results confirmed previous result that overexpression of Bcl-2 in 3T3 and C3H cell lines could enhance G0 quiescence and delay S phase entry by p27 stabilization.Citation16 To better understand the mechanisms of Bcl-2s cell cycle delay function, we investigated the role of mitochondrial ATP and ROS on cell cycle. In present study, ATP levels increased with time during re-stimulation for both Bcl-2 cells and PB controls. ATP peak was lower and occurred later in Bcl-2 cells than in PB cells, correlating with S phase delay in Bcl-2 cells, suggesting that ATP is an important mediator of Bcl-2 function. Our findings that changing the level of ATP resulted in cell cycle alteration further verified the regulatory role of ATP. ATP was reported as an extracellular messenger elevated in the tumor microenvironment, and exposure to high concentrations of ATP might promote cell cycle progression and cancer transformation. Lower ATP in Bcl-2 cells might be an important causative factor in delay of cell cycle and inhibition of cancer cell proliferation. Decreasing ATP might be a tumor suppressing strategy.
Our results showed that ROS levels were decreased in synchronized cells compared with normal growing cells. In addition, ROS levels were significant lower in synchronized Bcl-2 cells than those in PB controls, suggesting that Bcl-2 might mediate cell growth through ROS signaling. ROS are a series of reactive oxygen species produced from aerobic respiration in mitochondrial oxidative phosphorylation. Previous reports indicated that cancer cells exhibit more oxidative stress, and ROS were required for cell proliferation and essential for tumorigensis.Citation18 ROS played a role in regulation of late G1/S phase transition.Citation25 Consistent with these findings, we found ROS were lower in Bcl-2 cells, and G1/S phase transition was delayed in Bcl-2 cells, which supported our hypothesis that as a ROS scavenger, Bcl-2 could lower the concentration of ROS, which up-regulates p27, and inhibits cell proliferation.
Exposure of Bcl-2 cells to H2O2 did not raise ROS levels and had no effect on cell cycle progression, further supporting Bcl-2s role as a ROS scavenger and implicating ROS as regulatory signal in cell proliferation. On the contrary, reduction of ROS level in PB cells by GSH delayed S phase entry and increased p27, which mimics the phenotype of Bcl-2 cells. Bcl-2 lacks sequence or structural features of known anti-oxidant or redox active proteins, suggesting that anti-oxidant effects of Bcl-2 are indirect. The most likely explanation is that Bcl-2 modulates mitochondrial bioenergetics to decrease production of ATP and accumulation of ROS, which in turn regulate p27 protein expression to control cell cycle.Citation26 These results confirmed that ROS participates in regulation of G0/G1 to S phase and ROS may serve as a signal to modulate p27 expression.
The recent wave of molecular targeted therapies has focused on BCL−2 antagonist venetoclax (ABT-199) in clinical trials in hematologic malignancies.Citation27 However, targeting common biologic properties of cancer cells could make the drugs widely applicable across a range of cancer subtypes. More cancer have been found to be associated with mitochondria dysfunction of ATP and ROS generation,Citation28,29 a recent study found that reduced mitochondrial oxphos caused cell arrest and cell death.Citation7 In conclusion, our results revealed that Bcl-2 delayed cell cycle entry by decreasing ROS and ATP levels, the intrinsic mitochondrial pathway represents a promising target for novel therapies. Targeting this pathway has the potential to alter the therapeutic landscape for a variety of cancers including but not limited to Bcl-2 over-expressing cancer cells.
Materials and methods
Cell lines and cell culture
Mouse embryo fibroblast NIH3T3 and C3H/10T1/2 clone8 (C3H) cell lines were purchased from Shanghai cell bank of the Chinese Academy of Sciences, both have surface-specific characteristics of contact inhibition. Human kidney epithelial 293T cell line was used to make virus. All cells were cultured with Dulbecco's Minimum Essential Medium (DMEM) (Hyclone, Logan, USA, SH30022.01B), supplemented with 10% Fetal Bovine Serum (FBS) (Hyclone, Logan, USA, SH30084.03) and 1% penicillin/streptomycin (Hyclone, Logan, USA, SV30010) at 37 °C in 5% CO2.
Virus packaging and stable transfection
Recombinant pBABEpuro-Bcl-2 vector, packaging plasmid pCL−Eco (Takara Co., NBP2–29540), and X-tremeGENE HP DNA Transfection Reagent (Roche, Basel, Swiss, 06366236001) were added into 10% FBS DMEM medium, mixed gently and incubated at room temperature for 20 min. The mixture was added into 293T cells and cultured for 48 hr, the supernatant containing pBABEpuro-Bcl-2 virus was collected and filtered using a 0.45 μm filter. NIH3T3 (3T3) and C3H cells were then infected with the PB vector or Bcl-2 virus for 48 hr, the stable expressing cells were screened by antibiotic G418 (Gibco-BRL, Carlsbad, USA, 10131035), and confirmed by Western blotting.Citation30
Detection of ATP and ROS during serum re-stimulation
Cells were synchronized at G0/G1 stage by contact inhibition (CI) and serum starvation (SS). After re-stimulation by re-plating or serum addition, cells were harvested every 2 hr starting from 8 hr to 20 hr, and stained with PI and analyzed for cell cycle by BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). The proportion of cells in G0/G1, S, and G2/M phases was calculated by C6 software.
For each time point, ATP level were measured using ATP assay kit (Beyotime, Jiangsu, China, S0026). Bioluminescence was detected on a fluorescence illuminometer Pi-102 (Hygiene, New York, USA) and cellular ATP content (nmol/L) was calculated by ATP standard, further normalized by BCA protein quantification assay kit (Beyotime, Jiangsu, China, P0012).Citation31
Cells were collected and re-suspended in PBS at 1 × 105 cells/mL, then incubated with the mitochondria-specific dye, MitoSOX Red (Invitrogen-Life Technologies, New York, USA, M36008), for 30 min at 37 °C, untreated cells served as negative control. ROS generation was analyzed using BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA).Citation32
Alteration of ATP by oligomycin A and ATP sodium salt
PB or Bcl-2 cells were synchronized to G0/G1 phase by serum starvation or contact inhibition, then these arrested cells were re-stimulated to enter S phase. To assess the role of ATP on cell cycle regulation, 0, 5 or 8 μg/mL of oligomycin A (mitochondrial ETC inhibitor, Sigma, St Louis, USA, SIGMA-75351) was added into arrested PB or Bcl-2 cells during re-stimulation to reduce ATP production, cells were collected at 0, 8, 12, 16 and 20 hr for each concentration of oligomycin A, cell cycle and cell death were analyzed by flow cytometry, ATP levels were detected by ATP assay kit. To increase ATP level, 0, 50, 100 or 200 μg/mL ATP sodium salt (Sigma, St Louis, USA, SIGMA-A7699) were added into arrested PB and Bcl-2 cells, followed by cell cycle profile analysis at 0, 8, 10, 12, 16 and 20 hr to evaluate the effects of ATP alteration on cell cycle progression.
Alteration of ROS by hydrogen peroxide and reduced glutathione
PB or Bcl-2 cells were synchronized to G0/G1 phase by serum starvation or contact inhibition, then these arrested cells were re-stimulated to enter S phase. To scavenge ROS, 0, 5, 10 and 20 mmol/L of reduced glutathione (GSH, Sigma, St Louis, USA, SIGMA-G6013) were added to PB cells during synchronization and re-stimulation, cells were harvested at 10, 12, 14, 16 hr, cell cycle progression were checked by flow cytometry, and ROS levels were determined by mitochondrial specific dye mitoSOX. To boost ROS level, 100 μmol/L of hydrogen peroxide (H2O2, Sangon, Shanghai, China, RCS02178) were supplied Bcl-2 cells. Treated cells and untreated cells were then collected at corresponding time points to investigate the effects of ROS change.
Western blot
PB and Bcl-2 cells were synchronized to G0/G1 phase by serum starvation or contact inhibition, then these arrested cells were re-stimulated to enter S phase. Cells were collected at 0, 8, 10, 12, 16, 20 hr during re-stimulation, normal growing cells were collected as control, cells were then washed 2 times with PBS, and ice cold RIPA lysis buffers (Beyotime, Jiangsu, China, P0013C) was added, incubated on ice for 20 min, and centrifuged at 12,000 rpm for 15 min at 4°C. The supernatant was collected, total protein was measured with BCA kit (Beyotime, Jiangsu, China, P0012) adjusted to 4 µg/µl, and stored at −80°C for later use. After denaturing, the proteins were separated using conventional gel electrophoresis of 12.5% SDS-PAGE, and transferred to PVDF membrane with 100V constant voltage for 2 h. The membrane was then blocked with 5% milk PBST solution 2 h, the primary antibody (at 1:1,000 dilution) was added and incubated in an ice box for overnight, then the membrane was washed with PBST for 5 min for 3 times. A secondary antibody (at 1:2,000 dilution) was added at room temperature for 2 h and washed with PBST for 5 min for 3 times, and detected with the ECL chemiluminescence kit (Thermo scientific, Boston, USA, 32106) by a fluorescent device. Antibodies against Bcl-2 (2876), and p27 (2552S) were purchased from Cell Signaling Technology Co. (CST, Boston, USA), and anti-tubulin antibody (AT819) was obtained from Beyotime Company.Citation33
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The present study was financially supported by grants from the National Natural Science Foundation of China 81360310, 31106237.
References
- Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science 1985; 229(4720):1390-3; PMID:3929382; http://dx.doi.org/10.1126/science.3929382
- Walensky LD. BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ 2006; 13(8):1339-50; PMID:16763614; http://dx.doi.org/10.1038/sj.cdd.4401992
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. BAX activation is initiated at a novel interaction site. Nature 2008; 455(7216):1076-81; PMID:18948948; http://dx.doi.org/10.1038/nature07396
- Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene 2008;27(50):6398-406; PMID:18955968; http://dx.doi.org/10.1038/onc.2008.307
- Vail ME, Pierce RH, Fausto N. Bcl-2 delays and alters hepatic carcinogenesis induced by transforming growth factor alpha. Cancer Res 2001; 61(2):594-601; PMID:11212255
- Murphy KL, Kittrell FS, Gay JP, Jager R, Medina D, Rosen JM. Bcl-2 expression delays mammary tumor development in dimethylbenz(a)anthracene-treated transgenic mice. Oncogene 1999; 18(47):6597-604; PMID:10597264; http://dx.doi.org/10.1038/sj.onc.1203099
- Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015; 126(11):1346-56; PMID:26186940; http://dx.doi.org/10.1182/blood-2015-01-621870
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014; 15(1):49-63; PMID:24355989; http://dx.doi.org/10.1038/nrm3722
- Vervloessem T, La Rovere R, Bultynck G. Antagonizing Bcl-2s BH4 domain in cancer. Aging 2015; 7(10):748-9; PMID:26525307; http://dx.doi.org/10.18632/aging.100828
- Koczor CA, Shokolenko IN, Boyd AK, Balk SP, Wilson GL, LeDoux SP. Mitochondrial DNA damage initiates a cell cycle arrest by a Chk2-associated mechanism in mammalian cells. J Biol Chem 2009; 284(52):36191-201; PMID:19840931; http://dx.doi.org/10.1074/jbc.M109.036020
- Fehér A, Ötvös K, Pasternak TP, Pettkó-Szandtner A. The involvement of reactive oxygen species (ROS) in the cell cycle activation (G0-to-G1 transition) of plant cells. Plant Signal Behav 2008; 3(10):823-6; PMID:19704510; http://dx.doi.org/10.1007/s10725-006-9152-0
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324(5930):1029-33; PMID:19460998; http://dx.doi.org/10.1126/science.1160809
- Murphy M. How mitochondria produce reactive oxygen species. Biochem J 2009; 417(1):1-13; PMID:19061483; http://dx.doi.org/10.1042/BJ2008138614
- Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson CM, Yang E. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J 2003; 22(20):5459-70; PMID:14532118; http://dx.doi.org/10.1093/emboj/cdg533
- Linette G P, Korsmeyer S J. Cross talk between cell death and cell cycle progression: BCL-2 regulates NFAT-mediated activation. Proc Natl Acad Sci U S A 1996; 93(18):9545-52; PMID:8790367
- Janumyan Y, Cui Q, Yan L, Sansam CG, Valentin M, Yang E. G0 function of BCL2 and BCL-xL requires BAX, BAK, and p27 phosphorylation by Mirk, revealing a novel role of BAX and BAK in quiescence regulation. J Biol Chem 2008; 283(49):34108-20; PMID:18818203; http://dx.doi.org/10.1074/jbc.M806294200
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 2010; 107(5):2037-42; PMID:20133848; http://dx.doi.org/10.1073/pnas.0914433107
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010; 107(19):8788-93; PMID:20421486; http://dx.doi.org/10.1073/pnas.1003428107
- Hockenbery DM, Oltvai ZN, Yin X-M, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993; 75(2):241-51; PMID:7503812; http://dx.doi.org/10.1016/0092-8674(93)80066-N
- Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science 1993; 262(5137):1274-7; PMID:7503812; http://dx.doi.org/10.1126/science.8235659
- Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans 2008; 36(Pt3):343-7; PMID:18481954; http://dx.doi.org/10.1042/BST0360343
- Mehmeti I, Lenzen S, Lortz S. Modulation of Bcl-2-related protein expression in pancreatic beta cells by pro-inflammatory cytokines and its dependence on the antioxidative defense status. Mol Cell Endocrinol 2011; 332(1-2):88-96; PMID:20933054; http://dx.doi.org/10.1016/j.mce.2010.09.017
- Scarfo L, Ghia P. Reprogramming cell death: BCL2 family inhibition in hematological malignancies. Immunol Lett 2013; 155(1-2):36-9; PMID:24095849; http://dx.doi.org/10.1016/j.imlet.2013.09.015
- Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol 2013; 5(2):pii:a008722; PMID:23378584; http://dx.doi.org/10.1101/cshperspect.a008722
- Havens CG, Ho A, Yoshioka N, Dowdy SF. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol Cell Biol 2006; 26(12):4701-11; PMID:16738333; http://dx.doi.org/10.1128/MCB.00303-06
- Susnow N, Zeng L, Margineantu D, Hockenbery DM. Bcl-2 family proteins as regulators of oxidative stress. Semin Cancer Biol 2009; 19(1):42-9; PMID:19138742; http://dx.doi.org/10.1016/j.semcancer.2008.12.002
- Gibson CJ, Davids MS. BCL-2 Antagonism to Target the Intrinsic Mitochondrial Pathway of Apoptosis. Clin Cancer Res 2015; 21(22):5021-9; PMID:26567361; http://dx.doi.org/10.1158/1078-0432.CCR-15-0364
- van den Heuvel L, Smeitink J. The oxidative phosphorylation (OXPHOS) system: nuclear genes and human genetic diseases. Bioessays 2001; 23(6):518-25; PMID:11385631; http://dx.doi.org/10.1002/bies.1071
- Walker JE, Dickson VK. The peripheral stalk of the mitochondrial ATP synthase. Biochim Biophys Acta 2006; 1757(8):286-96; PMID:16697972; http://dx.doi.org/10.1016/j.bbabio.2006.01.001
- Chen G, Dai J, Tan S, Meng S, Liu Z, Li M, Cui Q, Yu M. MTERF1 regulates the oxidative phosphorylation activity and cell proliferation in HeLa cells. Acta Biochim Biophys Sin 2014; 46(6):512-21; PMID:24777141; http://dx.doi.org/10.1093/abbs/gmu029
- Xiong W, Jiao Y, Huang W, Ma M, Yu M, Cui Q, Tan D. Regulation of the cell cycle via mitochondrial gene expression and energy metabolism in HeLa cells. Acta Biochim Biophys Sin 2012; 44(4):347-58; PMID:22343378; http://dx.doi.org/10.1093/abbs/gms006
- Hahm ER, Sakao K, Singh SV. Honokiol activates reactive oxygen species-mediated cytoprotective autophagy in human prostate cancer cells. Prostate 2014; 74(12):1209-21; PMID:25043291; http://dx.doi.org/10.1002/pros.22837
- Fu XF, Yao K, Du X, Li Y, Yang XY, Yu M, Li MZ, Cui QH. PGC-1α regulates the cell cycle through ATP and ROS in CH1 cells. J Zhejiang Univ Sci B 2016; 17(2):136-46; PMID:26834014; http://dx.doi.org/10.1631/jzus.B1500158