
ABSTRACT
Cellular transitions are achieved by the concerted actions of regulated degradation pathways. In the case of the cell cycle, ubiquitin mediated degradation ensures unidirectional transition from one phase to another. For instance, turnover of the cell cycle regulator cyclin B1 occurs after metaphase to induce mitotic exit. To better understand pathways controlling cyclin B1 turnover, the N-terminal domain of cyclin B1 was fused to luciferase to generate an N-cyclin B1-luciferase protein that can be used as a reporter for protein turnover. Prior studies demonstrated that cell-based screens using this reporter identified small molecules inhibiting the ubiquitin ligase controlling cyclin B1-turnover. Our group adapted this approach for the G2-M regulator Wee1 where a Wee1-luciferase construct was used to identify selective small molecules inhibiting an upstream kinase that controls Wee1 turnover. In the present study we present a screening approach where cell cycle regulators are fused to luciferase and overexpressed with cDNAs to identify specific regulators of protein turnover. We overexpressed approximately 14,000 cDNAs with the N-cyclin B1-luciferase fusion protein and determined its steady-state level relative to other luciferase fusion proteins. We identified the known APC/C regulator Cdh1 and the F-box protein Fbxl15 as specific modulators of N-cyclin B1-luciferase steady-state levels and turnover. Collectively, our studies suggest that analyzing the steady-state levels of luciferase fusion proteins in parallel facilitates identification of specific regulators of protein turnover.
Introduction
Ubiquitin mediated degradation pathways play essential roles in cellular homeostasis.Citation1 In addition, many ubiquitin mediated proteasome pathway components are the targets of small molecule inhibitors used for the treatment of various cancers.Citation2 Therefore, a deeper understanding of interactions in the ubiquitin proteasome pathway is needed for understanding cell proliferation, organismal growth, as well as drug discovery in the ubiquitin pathway. A challenge for identifying these interactions, however, is that many are low-affinity and labile, thus making them difficult to uncover using traditional co-immunoprecipitation strategies. To circumvent this issue, alternative methods have been devised to use high throughput screening to identify upstream regulators of substrates in the ubiquitin proteasome pathway.Citation3,4 Our earlier studies modified a strategy used by King and colleagues to uncover small molecules controlling N-cyclin B1-luciferse turnover.Citation5 Utilizing a cell based screening approach where we expressed Wee1-luciferase in HeLa cells and incubated the transfected cells with small molecule inhibitors, we discovered kinase inhibitors that were selective for Casein kinase (CK)1.Citation6,7 Using both biochemical and genetic studies we then demonstrated that CK1 and GSK3 phosphorylate Wee1 to target it for destruction.Citation6,7 In a similar manner, using a p27Kip1-luciferase fusion construct we demonstrated that PKCα phosphorylates p27Kip1 and affects its degradation rate.Citation8 One of the main conclusions from these studies is that parallel screening of luciferase fusion proteins incubated with small molecules can uncover upstream modulators of a specific substrate's turnover.
In the present study we determined if parallel screening of luciferase fusion proteins could be adapted for overexpression screens using cDNAs. We used N-cyclin B1-luciferase, which we co-transfected with approximately 14,000 cDNAs into HeLa cells. We identified previously characterized regulators of cyclin B1-turnover including Cdh1 and Emi-1. In addition, we found that the F-box protein Fbxl15 similarly reduces N-cyclin B1-luciferase levels specifically. As Fbxl15 has been described as having a cell cycle role, we postulate that our studies provide proof-in-principle that this quantitative approach is useful for identifying upstream components of cyclin B1 degradation. We further suggest that this cell based screening approach can be expanded to other proteins degraded by the ubiquitin pathway.
Methods
cDNA library collection and preparation of screening sets
Approximately 5,500 human and 9,000 mouse full-length cDNAs cloned into pCMV-Sport6 vector (Invitrogen) generated by the Mammalian Gene Collection (MGC) were purchased from Open Biosystems. cDNA plasmids were prepared as described previouslyCitation9 with modifications. Bacterial stocks containing individual plasmids were inoculated into 96-well deep well culture blocks containing 1.5 ml terrific broth (TB)/ampicillin with a 96 pin hand tool (V&P Scientific, Inc.) and grown in a HiGro incubator/shaker (Gene Machines) at 37°C for 16–24 hours. Bacterial cultures were then spun down and media aspirated. Plasmid DNAs were prepared from bacteria with the NucleoSpin Robot-96 Plasmid kit (Macherey-Nagel) and Microlab Star robot (Hamilton Company). Purified DNA plasmids were quantified with the Spectramax 2 Spectrophotometer (Molecular Devices) and normalized to 25 ng/ml with the Microlab Star. Approximately 40 ng of prepped DNA was aliquoted into individual wells of 384-well bioassay plates (Greiner Bio-One) using a Minitrack 8 HTS robotic liquid handling system (Perkin Elmer).
cDNA library screening for activators of N-cyclin B1-luciferase degradation
Screening to identify library cDNAs that alter N-cyclin B1-luciferase or luciferase activity from the N-cyclin B1-luciferase or pGL3-Control reporter (Promega) was performed as follows: 40 ng of empty Sport6, pcs2-myc-Cdh1 or pcs2-myc-Emi1 plasmids was aliquoted into designated control wells of the 384-well library screening plate. 20 µl of a mixture containing 40 ng of either N-cyclin B1-luciferase or pGL3-Control and 45 ng of pCMV-Sport6 plasmids, 0.38 ml of TransIT-LT1 (Mirus Bio) transfection reagent, 20 µl of Optimem I (Invitrogen) were dispensed into each well with a Multi-Drop robot (TiterTek) and incubated at room temperature for 30–60 min. Subconfluent HeLa cells (ATCC) were trypsinized, harvested, and resuspended in fresh DMEM/20% FBS/Pen/Strep at a concentration of 5 × 105 cells/ml. After the 30–60 min incubation, 20 µl of HeLa cells were dispensed into each well with the Multi-Drop, and plates were incubated at 37°C for 24 hr. After 24 hr, 40 µl of BriteLite luciferin reagent (Perkin Elmer) was added to each well with the Multi-Drop, incubated for 1 min at room temperature, and luminescence levels quantified with an Analyst GT multimode reader (Molecular Devices). All screens were performed in duplicate.
Identification of activators of N-cyclin B1-luciferase degradation hits from primary screening and validation screening
Individual luciferase activities from each well (including controls) were normalized relative to the average activity from the population of library cDNAs (not including controls) within each plate. Average normalized activity and standard deviations were calculated for duplicate screening sets. Co-transfected cDNAs that caused 2-fold higher or lower changes in pGL3-Control activity relative to empty Sport6 vector were discarded. To identify cDNAs that specifically enhanced degradation/expression of N-cyclin B1-luciferase, 47 cDNAs that conferred a ratio of pGL3-Control normalized activity:N-cyclin B1-luciferase normalized activity ≥ 2.0 were hitpicked, prepped in 96-well format as described above, and 40 ng of each cDNA plasmid was spotted in quadruplicate within a 384-well plate. Hitpicks were validated for activity by performing reverse transfections and luciferase assays in triplicate with N-cyclin B1-luciferase, Wee1-luciferase, and pGL3-Control (luciferase alone) reporters as above.
Cycloheximide degradation assay
Subconfluent HeLa cells (ATCC) were transfected using Mirus Bio Transit-LT1 in 6-well dishes. 2.4 µg pGL3 construct / 2.4 µg flag or myc construct / 22.5 µl transfection reagent in 1.5 ml was added to 1.5 ml HeLa at 8 ×105 and allowed to grow for 48 hr. Cells were trypsinized and resuspended in 1X HeLa media at a concentration of 1.6 ×106 cells/ml. 50 µl of cells was added to 4 rows of wells of a 96-well white TC plate. Each row was in quadruplicate for each construct. 50 µl of 200 µg/ml cyxloheximide was added to the wells at 0, 1, 2 and 4 hr after plating. Immediately following the 4 hr cycloheximide treatment, 100 µl of Britelite was added to all the wells and RLU (Relative Luminescence Units) determined as above.
Mass spectrometry analyses
Anti-Flag-immunoprecipitations were performed on extracts from HEK-293T (ATCC) or HEK-293F (Invitrogen) transfected with Flag-Fbxl15, Flag-ΔN64-Fbxl15, or Flag alone and then gels were silver stained using Pierce's Silver SNAP Stain for Mass Spectrometry kit. Protein bands were excised, digested, and analyzed using liquid chromatography mass spectrometry. Briefly, excised bands were reduced and alkylated before trypsin digestion. Following digestion the peptides were extracted and dried. Digestion mixtures were reconstituted in 0.1 M acetic acid, loaded onto precolumns (360 mm o.d. × 100 mm i.d. fused silica, Polymicro Technologies) packed with 3 cm irregular C18 (5–15 cm non-spherical, YMC, Inc.) and washed with 0.1 M acetic acid for 5 min before switching in-line with the resolving column (7 cm spherical C18, 360 mm o.d. × 100 mm i.d. fused silica). Once the columns are in-line, the peptides were eluted along a gradient from 2% to 80% acetonitrile in 0.1 M acetic acid (flow rate of ∼350 nL/min). All samples were analyzed using a Thermo Electron LTQ. Electrospray was accomplished using a pulled fused silica emitter tip of approximately 5 cm with a voltage of 1.7 kV. The mass spectrometer was operated in data dependent mode with the top 5 most abundant ions in each spectrum selected for sequential MS/MS experiments. The exclusion list was used (1 repeat, 180 sec return time) to increase dynamic range. All MS/MS spectra were searched with Sequest (version 2.7) using sample dependent databases. Searches were performed with a fixed carbamidomethylation of 19 cysteine (C) and variable oxidation of methionine (M) and phosphorylation of serine (S) and threonine (T). Database search results were tabulated and visually inspected for correct assignment using Scaffold version 1.7 (Proteome Software).
FACS analyses
Samples were prepared as described.Citation10 Briefly, cells were harvested and centrifuged 500 × g for 5 min at 4°C. Cells were then washed with cold PBS, centrifuged 500 × g for 5 minutes at 4°C, and rapidly resuspended with 70% Ethanol. Cells were then centrifuged at 400 × g for 10 min at 4°C washed with cold PBS. The supernatant was removed, and the cell pellet was resuspended in 38 mM sodium citrate with 69 mM of propidium iodide and 19 mg/mL of RNAse A. FACS was performed with a BD Bioscience LSR II system and analyzed using Flowjo 8.7.3 software.
Western analyses
Cells were collected, centrifuged at 500 × g for 5 min at 4°C and rinsed with cold PBS and centrifuged again. The cell pellet was resuspended with Laemmli sample buffer containing 10 mM DTT. Samples were then sonicated, heated at 90°C for 10 min and loaded on 4–20% SDS–Page gel. Gels were transferred to 0.2-µM nitrocellulose membranes. Primary antibodies were purchased from Santa Cruz Biotechnology: Lamin A (H-102): sc-20680, Skp1 p19 (H–63): sc–7163, c-Myc (A-14):sc-789, cyclin B1: sc-245, p-histone H3-serine 10: sc-8656-R. Monoclonal Mouse Anti-FLAG M2 primary antibody was purchased from Sigma, Inc. Fbxl15 antibody was made by KLH-conjugated peptide CSLSRLRKRGVDIDV injection in rabbit using Zymed. Western blots were detected with Amersham ECL Plus Western blotting detection reagents (secondary antibody anti–rabbit IgG horseradish peroxidase conjugated).
Skp1 interactions
HEK-293F cells were transfected following the FreeStyle™ 293 Expression System manual (Invitrogen) with pCS2-msflg-HsFbxl5, pCS2-msflg-ΔN64HsFbxl5, or nothing. After 48 hr, extracts were made as describedCitation11 (Swelling buffer = 20mM Hepes, pH 7.7, 5mM MgCl2, 5mM KCl, 1µM Microcystin, and protease inhibitor cocktail from Sigma). EZview™ Red ANTI-FLAG® M2 Affinity Gel (St. Louis, MO) was washed 2X with swelling buffer and incubated with extract end-over-end, overnight at 4°C. 10% of the extract was saved for western analysis. The beads were then washed 2 times (each wash is 10% of bed volume) with wash buffer containing 0.2% Igepal, 2 times with wash buffer containing 300mM KCl, then twice with normal wash buffer (Wash buffer = 20mM Hepes, pH 7.7, 5mM MgCl2, 100mM KCl, and 10% glycerol). Proteins were eluted from the beads with Laemmli sample buffer and loaded on a gel for either western blot or silver stain.
Results
A cell based means of identifying cyclin B degradation regulators
We used a cell-based approach to identify proteins whose activities resemble the APC/C activator Cdh1. Since Cdh1 overexpression induces the degradation of cyclin B1,Citation12 we reasoned that a Cdh1-like protein would similarly reduce the steady-state level of cyclin B1. One measure of Cdh1 activity is N-cyclin B1-luciferase degradation, which has been used as an accurate probe both in extracts and cells.Citation13,5 For example, overexpression of known APC/C activators (Cdh1) or inhibitors (Emi-1) controls N-cyclin B1-luciferase levels in a fashion akin to their ability to regulate cyclin B1 levels via the APC/C.Citation14,15,13 We thus used N-cyclin B1-luciferase in a screen to identify cDNAs that function similar to Cdh1. Cells were transfected with N-cyclin B1-luciferase and either control vector, Cdh1, Emi-1, or 14,000 individual cDNAs whose concentration was normalized on 384-well plates. In an identical counterscreen, cells were transfected with control vector, Cdh1, Emi-1, or the normalized cDNAs with luciferase alone. Transfected cells were then incubated for 24 hours before adding Britelite reagent, which allows detection of luminescence signal and N-cyclin B1-luciferase or luciferase levels. (Supplemental Figure S1, Supplemental Table S1).
By comparing the signal generated from N-cyclin B1-luciferase-expressing cells to those generated from luciferase-expressing cells, we identified cDNAs whose expression specifically reduced N-cyclin B1-luciferase levels, expressed as a ratio of N-cyclin B1-luciferase to vector control (, , Fig. S1, Table S1). Importantly, Cdh1, when included in this screening set (labeled Cdh1 [S]) or added as control (Cdh1 [C]), specifically repressed N-cyclin B1-luciferase levels, whereas Emi-1 increased steady-state levels of N-cyclin B1-luciferase (, Fig. S1, Table S1). Interestingly, the F-box protein, Fbxl15 was similar to Cdh1 in that it specifically reduced N-cyclin B1-luciferase levels (, , Fig. S1, Table S1). Further, like Cdh1, Fbxl15 overexpression reduced the steady-state levels of N-cyclin B1-luciferase but not Wee1-luciferase or luciferase alone (, Fig. S1, Table S1). By contrast, other cDNAs such as Fbxl30 or Zip4 did not reduce the steady-state levels of N-cyclin B1-luciferase specifically (, Table S1). Fbxl15 induced N-cyclin B1-luciferase degradation yet did not affect steady-state levels of p27-luciferase, p21-luciferase or Wee1-luciferase in a comparative screen of 368 ubiquitin pathway cDNAs (Table S2). Fbxl15-mediated reduction of N-cyclin B1-luciferase was not due to effects on the cell cycle as FACS analyses demonstrated that Fbxl15 overexpressing cells had identical profiles as cells expressing vector alone (Fig. S1). The putative Fbxl15 Drosophila ortholog jetlag,Citation16 however, did not affect N-cyclin B1-luciferase levels, suggesting that a domain necessary for mediating this turnover is not conserved between the Drosophila and vertebrate proteins (Fig. S1).
Figure 1. Cdh1 and Fbxl15 specifically reduce N-cyclin B1-luciferase levels. A. Fbxl15 or Cdh1 overexpression reduces steady-state levels of N-cyclin B1-luciferase, but not Wee1-luciferase or Luciferase. HeLa cells were transfected with either N-cyclin B1-luciferase, Wee1-luciferase or luciferase alone along with either Fbxl15 or Cdh1, or other cDNAs listed. Steady-state luciferase activity was calculated relative to sport-6 vector alone control. All experiments were performed in triplicate. B. Cdh1 and Fbxl15 overexpression reduces steady-state levels of N-cyclin B-luciferase. Other cDNAs that were overexpressed such as Fbx30 or Zip4 did not affect steady-state levels of any luciferase fusion proteins.
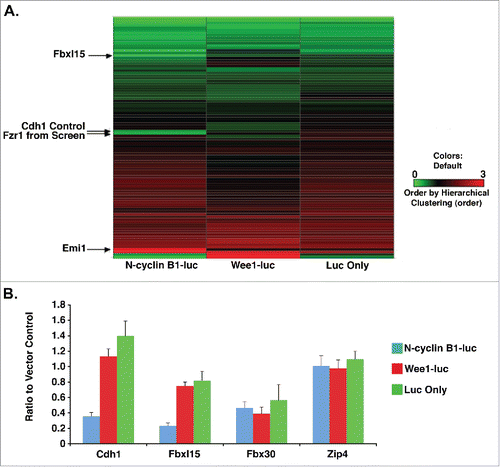
Figure 2. N-cyclin B1-luciferase degradation is accelerated in cells overexpressing Fbxl15 or Cdh1. A. Fbxl15 or Cdh1 overexpressing cells accelerate N-cyclin B1-luciferase turnover. Left Panel: HeLa cells were transfected with either N-cyclin B1-luciferase or luciferase alone along with vector, Fbxl15, or Cdh1. Transfected cells were then incubated for 48 hr after which time cycloheximide was added for the indicated times. Cells were then processed for luminescence signal after 0, 1, 2, or 4 hr. Right Panel: Representative Western blot demonstrating equal levels of Fbxl15 and Cdh1 in HeLa cell transfections. Loading control indicates non-specific band. B. Fbxl15 or Cdh1 overexpression does not cause global changes in cell cycle. HeLa cells were transfected with either eGFP-Fbxl15, eGFP, or Fbxl15, Cdh1, or vector cotransfected with eGFP and the number of eGFP+ cells in each cell cycle phase determined by PI-FACS analysis. All experiments were performed in triplicate.
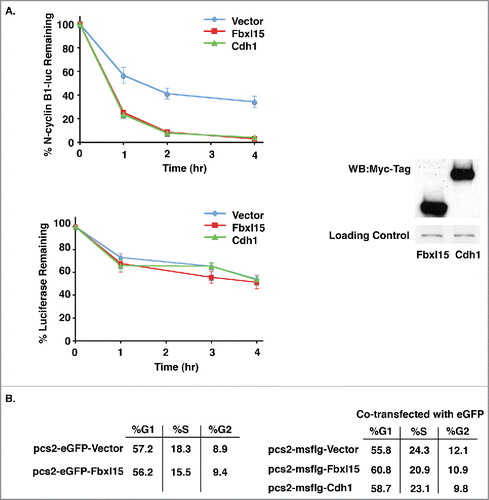
Fbxl15 specifically binds to Skp1
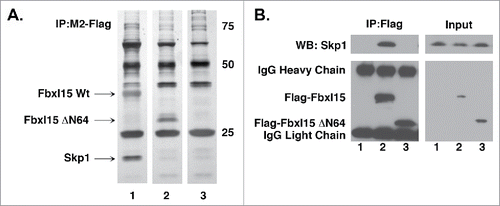
Based on its similarity to well-characterized F-box proteins, we suspected that Fbxl15 contains at least 2 domains required for its activity, an N-terminal Skp1 binding region and a C-terminal leucine-rich domain required for mediating protein-protein interactions.Citation17 Since loss of the Fbxl15 N-terminus disabled its ability to induce N-cyclin B1-luciferase turnover in multiple cell types (Fig. S2), we assessed whether this region indeed contained an F-box domain critical for binding Skp1. Immunoprecipitation/mass spectrometry (LC-MS/MS) as well as Western blot analyses of wild-type versus ΔN64-Fbxl15-expressing cells demonstrated that Skp1 bound to Fbxl15 and that this interaction required the N-terminus of Fbxl15 (, Figs. S3, S5). These studies are in keeping with a previously reported requirement for the N-terminal domain of Fbxl15 for binding Skp1.Citation18,19
Figure 3. Fbxl15 N-terminus contains a Skp1 binding domain A. The N-terminus of Fbxl15 is required for associating with Skp1. Silver-stained gel of anti-Flag immunoprecipitates of 293T cells transfected with either wild-type Flag-Fbxl15, Flag-ΔN64-Fbxl15 or Flag alone. The identity of the bands was confirmed by LC-MS/MS. B. Flag-Fbxl15 associates wth Skp1. 293T cells were transfected with wild-type Flag-Fbxl15, Flag-ΔN64-Fbxl15 or Flag alone along and the extent of endogenous Skp1 association determined after anti-Flag immunoprecipitation. All experiments were performed in triplicate.
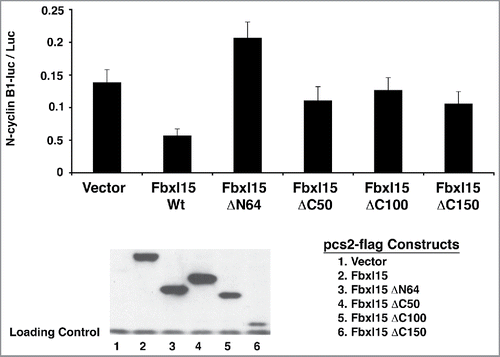
The N- and C-terminal domains of Fbxl15 are required for reducing the steady-state levels of N-cyclin B1-luciferase
To determine domains of Fbxl15 that mediate its ability to promote degradation of N-cyclin B1-luciferase, a series of N- and C-terminal truncation mutants were generated. Deletion of the N-terminus of Fbxl15 severely impaired its ability to reduce N-cyclin B1-luciferase levels (). Similarly, the Fbxl15 C-terminus was also required for reducing N-cyclin B1-luciferase levels (). Thus, the N- and C-terminal domains of Fbxl15 are required for regulating cyclin B1 levels. Although given the reduced expression of the C150 mutant, it is difficult to accurately assess the contribution of the C-terminus of Fbxl15 to cyclin B1 turnover. Collectively these studies suggest that Fbxl15 stimulates N-cyclin B1-luciferase turnover in an N- and C-terminal dependent manner.
Figure 4. Fbxl15 activity on N-cyclin B1-luciferase requires a functional N-terminus and C-terminus. Wild-type Fbxl15 or the indicated mutants were transfected with N-cyclin B1-luciferase for 24 hr and the amount of luminescence was determined. Lower panel: A representative Western blot of either Flag epitope-tagged wild-type Fbxl15 or the indicated mutants from transfected cells assessed for luciferase activity. Loading control indicates non-specific band. All experiments were performed in triplicate.
Discussion
We describe a quantitative high-throughput screen for the identification of regulators of N-cyclin B1-luciferase, a reporter of APC/C activity. This screen is applicable to multiple cell types (Supplemental Figure S2), and though the N-cyclin B1-luciferase screen biases for identifying “Cdh1-like” molecules, it is easily adapted for finding novel proteins whose biochemical activities resemble any component of a signaling or checkpoint pathway. Indeed, we have used this same strategy using p21Cip1-, p27Kip1-, and Wee1-luciferase to identify putative activators of p21Cip1, p27Kip1, and Wee1 turnover (Table S2). Interestingly, the regulators identified in these screens are specific for each luciferase fusion protein in the same manner that Cdh1 and Fbxl15 overexpression specifically reduced the steady-state levels of N-cyclin B1-luciferase. This apparent specificity may be due to our normalization procedure where we determined the ratio of protein-luciferase fusion protein to luciferase alone in each cell transfected with a given cDNA (Table S2).
Fbxl15 is similar to Cdh1 in stimulating N-cyclin B1-luciferase degradation (). Fbxl15's cell cycle role may be during interphase, mitosis, or both since it is expressed during the entire cell cycle and thus could target cyclin B1 for destruction during G1, S, or G2/M phases (Fig. S4). Consistent with this notion, Fbxl15 overexpression reduced N-cyclin B-luciferase levels in both mitotic and interphase cells (Fig. S5). Since a prior report demonstrated that Fbxl15 depletion in Hela cells reduced progression through the cell cycle,Citation18,19 it is feasible that the effects we have observed for Fbxl15 are merely due to redistribution of cells in different cell cycle phases after overexpression. However, at least in global FACS analysis, cells overexpressing Fbxl15 have the same cell cycle distribution as control transfected cells.
We acknowledge that one limitation of this cell based screening approach is that overexpression of cell cycle regulators can enrich for cell cycle phases where the protein fusion is expressed in high or low amounts. However, this expression would have to be at the post-transcriptional level since the protein-fusion is expressed from a constitutive promoter.
Fbxl15 has been characterized as a regulator of BMP signaling.Citation18,19 Given that the APC/C is activated by BMP signaling, we postulated that Fbxl15 might activate the APC/C by increasing BMP signaling. However, Fbxl15 knockdown did not affect BMP signaling in cerebellar granule cell progenitors (Fig. S6), a system where the APC/C is active.Citation20-22 Therefore, there might be other signaling pathways that Fbxl15 regulates that impinge on the APC/C and cyclin B1. Alternatively, Fbxl15's role in cell cycle progression might indirectly affect cyclin B1 turnover. We did not find that including recombinant Fbxl15 in cell-free extracts stimulated cyclin B1 turnover (unpublished observations). This may be due to a factor that is missing in our recombinant protein preparation. Future studies are required to delineate the role of Fbxl15 in cell cycle progression and cyclin B1 turnover and to adapt this cell based screening approach for other ubiquitin pathway substrates.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
NGA, TKA, and SS devised and executed the genome-wide screen. VC and JCB performed LC-MS/MS on Fbxl15 interacting proteins. NGA and SS performed all cell-based and biochemical experiments and wrote the paper. MEM performed all siRNA knockdowns of Fbxl15 in granule cell progenitors. NGA, TKA, SS, and MEM wrote the paper.
1301333_Supplemental_Material.zip
Download Zip (22.8 MB)Acknowledgements
We would like to thank all members of the Cancer Biology Department and Cell Based Screening Facility at Scripps Florida for helpful discussions and advice as well as members of the Center for Therapeutic Innovation and Lemmon-Bixby laboratory at the University of Miami.
Funding
This work was partly funded by a grant from the Landerberger foundation, and NS067289 to NGA.
Related Research Data
References
- Teixeira LK and Reed SI. Ubiquitin ligases and cell cycle control. Annu Rev Biochem 2013; 82:387-414; PMID:23495935; http://dx.doi.org/10.1146/annurev-biochem-060410-105307
- Penas C, Ramachandran V, and Ayad NG. The APC/C Ubiquitin Ligase: From Cell Biology to Tumorigenesis. Front Oncol 2011; 1:60; PMID:22655255
- Emanuele, MJ, Elia AE, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen YN, Rush J, Hsu PW, et al. Global identification of modular cullin-RING ligase substrates. Cell 2011; 147(2):459-74; PMID:21963094; http://dx.doi.org/10.1016/j.cell.2011.09.019
- Yen, HC and SJ Elledge Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science 2008; 322(5903):923-9; PMID:18988848; http://dx.doi.org/10.1126/science.1160462
- Verma R, Peters NR, D'Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, et al. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 2004; 306(5693):117-20; PMID:15459393; http://dx.doi.org/10.1126/science.1100946
- Penas C, Govek EE, Fang Y, Ramachandran V, Daniel M, Wang W, Maloof ME, Rahaim RJ, Bibian M, Kawauchi D, et al. Casein kinase 1delta is an APC/C(Cdh1) substrate that regulates cerebellar granule cell neurogenesis. Cell Rep 2015; 11(2):249-60; PMID:25843713; http://dx.doi.org/10.1016/j.celrep.2015.03.016
- Penas C, Mishra JK, Wood SD, Schürer SC, Roush WR, Ayad NG. GSK3 inhibitors stabilize Wee1 and reduce cerebellar granule cell progenitor proliferation. Cell Cycle 2015; 14(3):417-24; PMID:25616418; http://dx.doi.org/10.4161/15384101.2014.974439
- Trojanowsky M. Vidovic D, Simanski S, Penas C, Schurer S, Ayad NG. Screening of cell cycle fusion proteins to identify kinase signaling networks. Cell Cycle 2015; 14(8):1274-81; PMID:25606665; http://dx.doi.org/10.1080/15384101.2015.1006987
- Chanda SK, White S, Orth AP, Reisdorph R, Miraglia L, Thomas RS, DeJesus P, Mason DE, Huang Q, Vega R, et al. Genome-scale functional profiling of the mammalian AP-1 signaling pathway. Proc Natl Acad Sci U S A 2003; 100(21):12153-8; PMID:14514886; http://dx.doi.org/10.1073/pnas.1934839100
- Smith A, Simanski S, Fallahi M, Ayad NG. Redundant ubiquitin ligase activities regulate wee1 degradation and mitotic entry. Cell Cycle 2007; 6(22):2795-9; PMID:18032919; http://dx.doi.org/10.4161/cc.6.22.4919
- Ayad NG, Rankin S, Ooi D, Rape M, Kirschner MW. Identification of ubiquitin ligase substrates by in vitro expression cloning. Methods Enzymol 2005; 399:404-14; PMID:16338372
- Bashir T. Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 2004; 428(6979):190-3; PMID:15014502; http://dx.doi.org/10.1038/nature02330
- Harmey D, Smith A, Simanski S, Moussa CZ, Ayad NG. The anaphase promoting complex induces substrate degradation during neuronal differentiation. J Biol Chem 2009; 284(7):4317-23; PMID:19047054; http://dx.doi.org/10.1074/jbc.M804944200
- Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 2001; 105(5):645-55; PMID:11389834; http://dx.doi.org/10.1016/S0092-8674(01)00361-0
- Fang G, Yu H, Kirschner MW Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol Cell 1998; 2(2):163-71.
- Koh, K, X Zheng, and A Sehgal JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science 2006. 312(5781):1809-12; PMID:PMID:16794082; http://dx.doi.org/10.1126/science.1124951
- Jin, J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 2004; 18(21):2573-80; PMID:15520277; http://dx.doi.org/10.1101/gad.1255304
- Cui, Y, He S, Xing C, Lu K, Wang J, Xing G, Meng A, Jia S, He F, Zhang L. SCFFBXL(1)(5) regulates BMP signalling by directing the degradation of HECT-type ubiquitin ligase Smurf1. EMBO J 2011; 30(13):2675-89; PMID:21572392; http://dx.doi.org/10.1038/emboj.2011.155
- Li, D, Xie P, Zhao F, Shu J, Li L, Zhan Y, Zhang L. F-box protein Fbxo3 targets Smurf1 ubiquitin ligase for ubiquitination and degradation. Biochem Biophys Res Commun 2015. 458(4):941-5; PMID:25721664; http://dx.doi.org/10.1016/j.bbrc.2015.02.089
- Kalinovsky, A, Boukhtouche F, Blazeski R, Bornmann C, Suzuki N, Mason CA, Scheiffele P. Development of Axon-Target Specificity of Ponto-Cerebellar Afferents. PLOS Biology 2011. 9(2):e1001013; PMID:21346800; http://dx.doi.org/10.1371/journal.pbio.1001013
- Patzke, H, Reissmann E, Stanke M, Bixby JL, Ernsberger U. BMP growth factors and Phox2 transcription factors can induce synaptotagmin I and neurexin I during sympathetic neuron development. Mech Dev 2001. 108(1–2):149-159; PMID:11578868; http://dx.doi.org/10.1016/S0925-4773(01)00503-2
- Su, H-L, Muguruma K, Matsuo-Takasaki M, Kengaku M, Watanabe K, Sasai Y. Generation of cerebellar neuron precursors from embryonic stem cells. Dev Biol 2006. 290(2):287-96; PMID:16406324; http://dx.doi.org/10.1016/j.ydbio.2005.11.010