
The promise of targeted therapeutics for a defined oncogenic molecular lesion is always tempered by the likelihood of acquired resistance to the pharmacological or biologic agent. A prominent mode of acquired resistance to targeted therapy is the activation of “bypass-track” signaling, alternative nodes that can overcome the initial drug insult and promote growth. This adaptation can manifest at the transcriptional level to boost the abundance of, for example, key mitogenic kinases or at the level of protein activation, whereby post-translational modifications (e.g., phosphorylation) of signaling molecules are actively modulated. Activation of the mitogen-activated protein kinase pathway, primarily through genomic amplification of pathway members upstream of extracellular signal-regulated kinase (ERK), commonly contributes to the growth of triple negative breast cancer (TNBC). Adaptive bypass signaling in TNBC in response to mitogen-activated protein kinase kinase (MEK) inhibition (MEKi) involves transcriptional upregulation of receptor tyrosine kinases (RTKs) and their cognate ligands, via c-Myc degradation, to reactivate ERK and drive resistance.Citation1
One strategy for combating the TNBC adaptive transcriptional response to MEK inhibition is kinase inhibitor polypharmacology, yet variability in the adaptive RTK cohort, lack of selective inhibitors, and acquired resistance to the cocktail all ultimately limit the durability of this approach. Thus, we and others have demonstrated that BET bromodomain inhibitors, targeting BRD2, 3, 4, are capable of synergizing with targeted agents by broadly attenuating the adaptive response as opposed to a strategy targeting a few selected kinases with multiple inhibitors.Citation2,Citation3 BRD4 reads chromatin through the binding of its tandem bromodomain modules to acetyl lysine residues of histones and regulates transcription by recruiting P-TEFb to release paused RNA polymerase.
Our recent work uncovered a new model of BET bromodomain inhibitor efficacy in extending the durability of MEK inhibitor-induced growth suppression in TNBC.Citation4 We observed robust transcriptional responses to MEK inhibition by trametinib in both TNBC patient tumors following a 7-day trametinib regimen and in preclinical TNBC cell line models. A potential mechanism for combination BET bromodomain inhibitor + MEK inhibitor synergy is a promoter-centric model whereby transcripts are induced in response to trametinib and concurrent BET bromodomain inhibition (BETi) displaces BRD4 solely from these trametinib-induced adaptive response promoters. In contrast, we found expansive, genome-wide and rapid (1–4 h) remodeling of the epigenomic landscape beyond that of promoters in response to trametinib. De novo formation of BRD4-rich enhancers and super-enhancersCitation5 was widespread, as well as remodeling of the existing enhancer landscape. Trametinib-induced enhancer remodeling included a population of enhancers that were decommissioned, having lost BRD4 and associated factor density, and suggestive of potential “recommissioning” of enhancer factor density ().Citation6 The newfound enhancers were co-occupied with the activating histone modification H3K27ac, and the lysine acetyltransferase p300 as well as with prototypical enhancer components mediator (MED1) and CCAAT-enhancer-binding protein β (CEBPB).
Figure 1. Epigenomic landscape modulation as adaptation to MEK inhibition. Following trametinib treatment, triple negative breast cancer cells adapt by seeding the formation of new BRD4-rich enhancers to induce transcription of proximal receptor tyrosine kinase genes and other genes in growth/survival promotion pathways leading to drug resistance. BET bromodomain inhibition attenuates this response in combination with MEK inhibition.
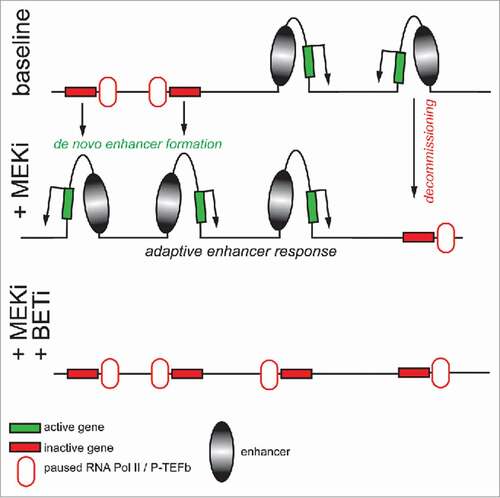
Co-treatment of trametinib with the BET bromodomain inhibitor JQ1 squelched the epigenomic landscape remodeling that occurred in response to trametinib, which correlated with suppression of transcripts of genes proximal to the trametinib-induced/JQ1-suppressed enhancer density and with transcripts of genes previously implicated in the adaptive response to MEKi. What was the functional culmination of combined MEKi/BETi in the context of TNBC? JQ1-mediated disruption of adaptive transcription following MEKi led to enhanced durability in long-term (4 week) colony formation assays, resensitization of resistant cells to trametinib, and synergized with trametinib for tumor growth inhibition in multiple TNBC mouse models.
An enhancer remodeling paradigm centered around BRD4 and its known association with the P-TEFb transcriptional elongation regulatory complex allowed us to posit targets in addition to BET bromodomains to combat the epigenomic rearrangements following MEK inhibition. Indeed, small molecule inhibition of core P-TEFb constituent CDK9, or BRD4-associated factors p300 or jumonji family demethylases abrogated adaptive RTK upregulation.
Are all TNBC cells equally adept in chromatin rewiring in response to targeted kinase inhibition? We used a mixed population TNBC cell line, SUM229-PE, containing an epithelial, EpCAM-positive population that possesses molecular characteristics of the basal-like intrinsic TNBC molecular subtype and a mesenchymal, EpCAM-negative population that has a gene expression signature that profiles as the claudin-low TNBC subtype. We found that the EpCAM positive population failed to remodel the BRD4 epigenome following trametinib treatment while the EpCAM negative population responded to trametinib with de novo enhancer formation to a similar degree as other claudin-low and basal-like TNBC cell lines used in the study. Could enhancer response underlie the differential sensitivity of the 2 populations to trametinib? Indeed, the EpCAM negative population in which we observed epigenomic remodeling was less sensitive to trametinib than the EpCAM positive population. Although the factors responsible for this distinction are currently unknown, this finding suggests different TNBC tumor cell populations may more effectively mount an enhancer response to drug leading to their selection during the onset of resistance.
Is the expansive chromatin remodeling that underlies the adaptive transcriptional response to trametinib an inherent or unique property of MEK inhibition? In addition to our study using an FDA-approved small molecule MEK inhibitor, genetic manipulation of MAPK members in fibroblasts has been shown to influence the enhancer landscape.Citation7 Inducible enhancers have been well-characterized in other stimulation contexts like cytokines in macrophages,Citation6 yet to what general extent other targeted therapeutics in cancer elicit an enhancer induction response is unknown. We suggest that determining enhancer responses using a diverse inhibitor library and different cell systems would be valuable for deciphering mechanisms of adaptive transcription leading to drug resistance. Transitioning from defining adaptive response chromatin landscapes in preclinical cell line models to in vivo murine models and with pre and post treatment patient tumors will be critical for achieving this goal.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012; 149:307-21; PMID:22500798; http://dx.doi.org/10.1016/j.cell.2012.02.053
- Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KAL, Granger DA, Reuther RA, et al. Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep 2015; 11:390-404; PMID:25865888; http://dx.doi.org/10.1016/j.celrep.2015.03.037
- Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, Parsons R. Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell 2015; 27:837-51; PMID:26058079; http://dx.doi.org/10.1016/j.ccell.2015.05.006
- Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU, et al. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacological targeting of the P-TEFb complex. Cancer Discov 2017; 7(3):302–321; PMID:28108460; http://dx.doi.org/10.1158/2159-8290.CD-16-0653
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013; 153:320-34; PMID:23582323; http://dx.doi.org/10.1016/j.cell.2013.03.036
- Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, Bair S, Newton G, Lichtman AH, Kung AL, et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 2014; 56:219-31; PMID:25263595; http://dx.doi.org/10.1016/j.molcel.2014.08.024
- Nabet B, Ó Broin P, Reyes JM, Shieh K, Lin CY, Will CM, Popovic R, Ezponda T, Bradner JE, Golden AA, et al. Deregulation of the Ras-Erk signaling axis modulates the enhancer landscape. Cell Rep 2015; 12:1300-13; PMID:26279576; http://dx.doi.org/10.1016/j.celrep.2015.06.078