
ABSTRACT
Cellular quiescence is a reversible cell growth arrest that is often assumed to require a persistence of non-permissive external growth conditions for its maintenance. In this work, we showed that androgen could induce a quiescent state that is self-sustained in a cell-autonomous manner through a “hit and run” mechanism in androgen receptor-expressing prostate cancer cells. This phenomenon required the set-up of a sustained redox imbalance and TGFβ/BMP signaling that were dependent on culturing cells at low density. At medium cell density, androgens failed to induce such a self-sustained quiescent state, which correlated with a lesser induction of cell redox imbalance and oxidative stress markers like CDKN1A. These effects of androgens could be mimicked by transient overexpression of CDKN1A that triggered its own expression and a sustained SMAD phosphorylation in cells cultured at low cell density. Overall, our data suggest that self-sustained but fully reversible quiescent states might constitute a general response of dispersed cancer cells to stress conditions.
Introduction
Cellular quiescence is a reversible growth arrest in the G0/G1 phase of the cell cycle that can be triggered by various conditions such as cell-contact inhibition, depletion of nutrient or growth factors, and exposure to growth-inhibitory cytokines or various chemicals. External conditions involved in the induction of the quiescent state are usually required for its maintenance, suggesting that cell quiescence is a passive phenomenon. At variance with this view, we recently described a more stable form of cell quiescence that was induced upon culturing prostate cancer cells at low cell density in a slightly hypertonic medium and which we designated as dormancy. A large fraction of the cells remained in the quiescent state when they were returned under growth permissive conditions in hypotonic or isotonic medium.Citation1 This suggested that the maintenance of this quiescent state depended mainly on cellular mechanisms that are self-sustained in the dormant cell population independently of the initial conditions that triggered it. However, we were not able to rigorously characterize the mechanisms involved in the self-sustaining of this quiescent state due to a high background level of dormancy escape when cells were returned under permissive growth conditions. We only showed that maintenance of cells under hypertonic conditions induced some redox imbalance and BMP/TGFβ signaling and that these signaling pathways fully accounted for the expression of a corresponding dormancy signature comprising 20 genes involved in cell cycle control, response to cellular stress, differentiation, stemness and BMP/TGFβ signaling.Citation2
Androgens are critically required for the proliferation of luminal epithelial stem cells, the differentiation and survival of their progeny in normal prostate and for the proliferation and survival of most early-stage prostate cancer cells.Citation3-5 Accordingly, the primary treatment of prostate cancer after radical prostatectomy is to inhibit AR signaling through depletion of circulating androgens and/or treatment with antagonists of AR.Citation5 However, this anti-androgen therapy eventually fails, with a median disease-free survival of a few years, due to the emergence of castration-resistant prostate cancer (CRPC) cells. In most cases, CRPC cells remain dependent on continued AR signaling for their survival and proliferation, the apparent independency from androgen being associated with genetic or epigenetic alterations enhancing AR activity under conditions of androgen ablation.Citation5 However, it has also been observed that androgens can inhibit proliferation of human prostate cancer cell in culture or/and growth of human prostate cancer cell xenografts in mouse.Citation6-8,9,10,11-17 This occurs for cells which are progressing toward androgen independence but are still expressing functional androgen receptors (AR) and at concentrations which can be within the range of physiologic androgen levels.Citation11,13,14,16 This has led to propose that androgens could constitute a complementary treatment of CRPC.Citation7,10-14,16
In the present work, we investigated the mechanisms of cell growth arrest that can be induced in prostate cancer cells upon treatments with androgens. We mainly used 2 androgen-responsive prostate cancer cell lines, LNCaP* and VCaP, that were routinely passaged in a growth medium supplemented with 10% fetal calf serum, that provided only low basal concentrations of androgens.Citation18 Of note, AR gene is amplified in VCaP cells which confers them a CRPC cell phenotype linked to an enhanced AR signaling pathway.Citation16,19 We showed that, when cultured at low cell density, transient exposure of these prostate cancer cells to low concentrations of androgens induced a self-sustained quiescent state. We demonstrated that this remarkable stability of quiescent state relied on a sustained redox imbalance and BMP/TGFβ signaling. These effects of androgens could be fully mimicked by transient overexpression of the oxidative stress marker CDKN1A that triggered its own expression and BMP/TGFβ signaling in cells cultured at low density.
Results
Androgen treatment of prostate cancer cells cultured at low density induces a G0/G1 cell growth arrest in an androgen receptor-dependent manner
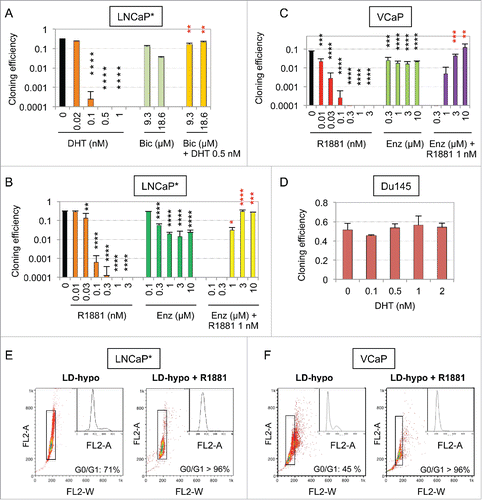
We observed that androgens, including testosterone, dihydrotestosterone (DHT) and R1881, a synthetic non-metabolizable androgen, strongly inhibited proliferation of LNCaP* prostate cancer cells plated at low cell density in a slightly hypotonic growth medium (LD-hypo) optimal for clonal cell proliferation.Citation1,2 Indeed, at doses of androgen superior to 0.3 nM cloning efficiency of these cells was decreased up to several hundred times (). Similar data were obtained with native LNCaP, or VCaP prostate cancer cells but not with Du145 cells lacking AR expression or 22Rv1 cells that constitutively expresses an AR isoform lacking the ligand-binding domain and displays a strongly attenuated induction of AR target genes by AR agonistsCitation11,20-22 (). This suggested that this growth-inhibitory effect was dependent on a functional AR. Accordingly, AR antagonists bicatulamide (Bic) or enzalutamide (Enz) strongly alleviated the growth inhibition mediated by androgens when these antagonists were used at clinically relevant concentrations (). Microscope examination of low cell density cultures suggested that, at concentrations between 0.2 and 0.5 nM, androgens induced a complete arrest of cell proliferation without conspicuous toxicity. Flow cytometry analysis of cell cycle upon propidium-iodide staining confirmed that more than 96% of the cells harvested after culturing at low density in the presence of R1881 for 7 d were confined to the G0/G1 phase of the cell cycle ().
Figure 1. Strong inhibitory effect of androgens on the cloning efficiency of AR-expressing prostate cancer cells and its suppression by AR antagonists. (A, B, C, and D) Cloning efficiency of LNCaP*, VCaP and Du145 prostate cancer cells in the presence of the indicated concentration of androgen and/or AR antagonist. Reported values are the mean ± sd derived from at least 2 independent experiments with duplicates. Asterisks indicate statistical significance of the growth-inhibitory effects of androgens as compared with non-treated cells (black asterisks) and of its suppression by AR antagonists as compared with cells treated only with androgen (red asterisks). (E and F) Cell cycle analysis of LNCaP* and VCaP cells cultured for 7 d under LD-hypo conditions in the presence of none or 0.5 nM R1881. Cells were analyzed by flow cytometry after propidium iodide staining. A 2-step gating procedure using Side SCattering-Height versus Forward SCattering-Height (data not shown) followed by pulse Area vs. pulse Width FLuorescence (FL2-A vs. FL2-W) was performed to eliminate cell doublets. Gated region is indicated on the FL2-A vs. FL2-W dot plot representation. Insets show fluorescence histograms. The percentage of cells in the G0/G1 phase of cell cycle is indicated. One representative experiment about 3 is shown.
Transient androgen treatment of prostate cancer cells at low cell density is sufficient to induce a self-sustained cellular quiescence (dormancy) that is reversed by BMP/TFGβ signaling inhibitors and reduced thiols
To get more insights into the mechanisms involved in androgen-induced growth-arrest, we analyzed whether androgen treatment of LNCaP* or VCaP cells cultured at low cell density could induce the expression of a dormancy signature comprising 20 genes, that we have previously characterized for prostate cancer cells rendered quiescent by culturing cells at low cell density in hypertonic medium.Citation2 A 7-day treatment with R1881 modulated expression of nearly all these dormancy signature genes with a significant induction of stress, differentiation and SMAD signaling markers (, S1, red bars). Since the dormant state elicited by hypertonicity displayed some stability, a large fraction of the cells remaining in a quiescent state after returning under hypotonic conditions,Citation1 we examined whether the growth-arrested state elicited by androgens shared similar properties. We observed that a transient treatment with R1881 at doses superior to 0.2 nM for 7 d was sufficient to induce a strong decrease of cloning efficiency in LNCaP* and VCaP cells (). A similar observation could be made with native LNCaP cultured under atmospheric (21%) or more physiologic (3%) oxygen concentration (Fig. S2 and data not shown). As cells were maintained in culture for 20 d after withdrawal of R1881 in our assay and 10 d were sufficient for non-treated cells to form identifiable cell clones, the strong decrease in cloning efficiency caused by transient R1881 treatment suggested that the cell growth-arrest induced by R1881 persisted at least 10 d after its withdrawal. Analysis of cell cycle by flow cytometry confirmed that more than 91% of the treated cells were still in the G0/G1 phase of the cell cycle 7 d after withdrawal of R1881 (). Additionally, RT-qPCR analysis cells performed 7 d after withdrawal of R1881 (i. e. 14 d after the initiation of experiment) showed that most of the dormancy signature genes remained expressed at levels closed to those measured just at the end of the androgen treatment at day 7 (). The slight decrease in dormancy genes expression observed at day 14 could result from the contribution of proliferating cells that have escaped dormancy induction and/or to the decrease of AR signaling that directly regulated transcription of genes like KLK3 (encoding Prostate Specific Antigen). Overall, our data suggested that the growth-arrest induced by transient androgen treatment at low cell density was stable after withdrawal of the exogenous androgens, at least on a time scale of 10 d. Nevertheless, this growth arrest could be efficiently reversed if a coktail of glutathione (GSH) or N-acetylcysteine (NAC) and inhibitors of TGFβ/BMP receptors (K02288 and SB505124) were added at the end of androgen treatment at day 7 of the experiment (GSK and NSK in and S2, pink bars). Interestingly, AR antagonists, in contrast to GSK cocktail, were ineffective to reverse the growth arrest induced by androgens when they were added at the end of R1881 treatment (at day 7, ). As a control, 7-day treatment with the sole AR antagonist Bic or Enz only slightly decreased cloning efficiency, indicating that the growth-inhibitory effect of AR antagonists, in contrast to AR agonists, was largely spontaneously reversed upon their withdrawal ( and see also for the effects of longer treatments). These experiments showed that the stability of the quiescent state induced by transient exposure to androgens did not rely on a sustained AR activity but rather was achieved through a Hit-and-Run mechanism.
Figure 2. Transient exposure of cells cultured at low density to androgens induced a self-sustained but reversible growth arrest in the G0/G1 phase of cell cycle. (A) Scheme of experimental design for transitory R1881 treatments. Cells were cultured for 7 d under LD-hypo conditions in the presence R1881 at the indicated concentration, then washed 3 times and further cultured in the presence of the indicated compounds (LD-hypo + R1881 → LD-hypo ± none or GSK or NSK). GSK: 8 mM GSH, 0.2 µM SB505124 and 0.2 µM K02288; NSK: 8 mM NAC, 0.2 µM SB505124 and 0.2 µM K02288. For cloning efficiency measurement, cell colonies were counted at day 27. RT-qPCR and flow cytometry analysis were performed at day 7 (LD-hypo and LD-hypo + R1881) and day 14 (LD-hypo + R1881 → LD-hypo). (B and C) RT-qPCR analysis of the variations in the expression of the dormancy signature genes according to the indicated cell culture conditions in LNCaP* and VCaP cells. R1881 was used at a 0.5 nM concentration. Values are the mean ± sd of 2 independent experiments. Asterisks indicated statistical significance for the differences in mRNA levels between cells cultured under LD-hypo and LD-hypo + R1881 (red asterisks) or LD-hypo + R1881 → LD-hypo (black asterisks) conditions. (D and E) Inhibition of the cloning of LNCaP* and VCaP cells by R1881 and its subsequent reversal by the GSK or NSK mix. Data are averaged from 2 and 3 independent experiments for LNCap and VCaP cells respectively. Statistical significance of the effect of R1881 compared with non-treated cells (red asterisks) and of the effects of the GSK or NSK mix compared with cells treated with R1881 only (blue asterisks) are indicated. (F and G) Cell cycle analysis of LNCaP* and VCaP cells after a 7-days culture in the presence 0.5 nM R1881 followed by a 7-days culture without R1881 under LD-hypo conditions. Data are from the same experiment as those displayed in .
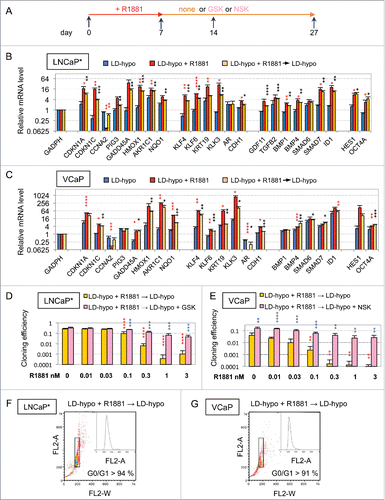
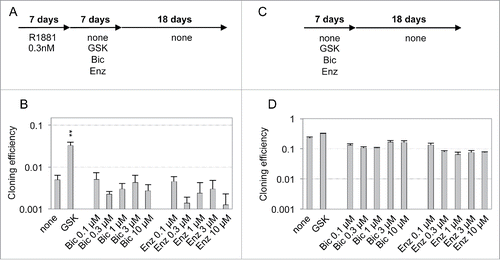
Figure 3. Ineffectiveness of AR antagonists to reverse dormancy established after a transient exposure to R1881. (A and C) Schemes of the experimental design for AR antagonist treatments. (B and D) Cloning efficiencies of the cells cultured as indicated. Values are the mean ± sd of 2 independent experiments. Only GSK (8 mM GSH, 0.2 µM SB50512 and 0.2 µM K02288) treatment displayed a significant effect on the reversal of androgen-induced dormancy (black asterisks).
Androgen-induced SMAD signaling and redox imbalance became self-sustained in dormant cells
The data described above suggested that androgens could induce a stable cellular quiescence (that we call dormancy from here) through the induction of both cell redox imbalance and SMAD signaling activation. Thus, we assessed SMAD phosphorylation in androgen-induced dormancy by Western blot. As shown in (see also ), SMAD phosphorylation was increased by the mere culture of VCaP or LNCaP* cells at low cell density for 7 days, in agreement with our observation that culture at low cell density is sufficient to increase expression of markers of TGFβ/BMP activation (, S1 and seeCitation2). R1881 treatment further increased SMAD phosphorylation level () and this was even more pronounced 7 d after R1881 withdrawal (at the 14th day of culture), suggesting that SMAD phosphorylation had become self-sustained in dormant cells (). Modulation of gene transcription by SMAD signaling was directly supported by analyzing the effects of its inhibition through expression of a small hairpin RNA targeting SMAD4 (shSMAD4Citation2). RT-qPCR analysis of the dormancy signature genes showed that SMAD4 depletion significantly decreased the ability of R1881 to enhance the expression of genes that are known targets of TGFβ/BMP-SMAD signaling such as ID1 or HES1 (Fig. S3 and see alsoCitation2). Of note, induction of oxidative stress markers like CDKN1A, HMOX1 and AKR1C1 was also blunted, suggesting a modulation of redox imbalance signaling by SMAD-mediated signaling pathway (Fig. S3 and see alsoCitation2). Finally, the functional involvement of TGFβ/BMP signaling in the growth arrest induced by androgens was evidenced by the strong reversal effect of the BMP receptor inhibitor K02288 (targeting ALK1, 2, 3, 6Citation23) on the dormant state induced by R1881 ( and S4). The TGFβ receptor inhibitor SB505124 (targeting ALK4, 5, 7Citation24) displayed lesser, if any, effects when used alone but reproducibly cooperated with K02288 to reverse dormancy ( and S4). Similarly, depletion of SMAD4 upon shSMAD4 expression led to a resistance of cells to the growth-inhibitory effect of androgens ( and S2), confirming the implication of SMAD signaling.
Figure 4. Implication of SMAD signaling and redox imbalance in the maintenance of the dormant state induced by transient exposure to the R1881 androgen. (A) Western blot analysis of the levels of SMAD phosphorylation and SMAD1, HMOX1 and CDKN1A expression in LNCaP* and VCaP cells under the indicated culture conditions (see also ). Numbers indicate the relative levels of GADPH or the relative amounts of the indicated proteins normalized by GAPDH levels. (B) Western blot analysis of the oxidation of the GRX1-roGFP2 sensor in LNCaP* cells under the indicated culture conditions. “red” and “ox” indicate reduced and oxidized forms of GRX1-roGFP2. Values in the lower panel indicate the ratio of oxidized to reduced GRX1-roGFP2 (ox/red). R1881 was used at a 0.6 nM concentration. The positive control, MD-hypo + H2O2, was cells cultured at medium cell density and treated with 100 mM H2O2 for 10 min before cell lysis. Values are mean ± sd derived from 3 independent transduced cell populations. Asterisks indicate the statistical significance of the variation of the ox/red ratio between MD-hypo and other cell culture conditions. (C and D) GSH supplementation blunted the gene expression signature of dormancy. Cells were cultured for 7 d in the presence of 0.2 nM R1881or 0.2 nM R1881 plus 8 mM GSH under LD-hypo conditions. Variations in the expression of the dormancy signature genes were measured by RT-qPCR as in . Values are mean ± sd of 2 independent cell culture experiments. Statistical significance of the differences between dormant and GSH-treated cells is indicated. (E) Partial suppression of androgen-induced dormancy by depletion of endogenous SMAD4 or by GSH supplementation. Cloning efficiency of shLuc (control) and shSMAD4-transduced cell populations were measured in non-treated cells (Ø) or in cells treated for 7 d with 0.3 nM R1881 followed by a culture for 20 additional days in growth medium with none (Ø) or with 8 mM GSH. Values are the mean ± sd derived from 2 experiments with 2 control- and 2 shSMAD4-transduced cell populations. Statistical significance of the variation between shSMAD4 and shLuc cells and of the effect of GSH is indicated by asterisks. (F and G) Reversal of R1881-induced dormancy in LNCaP* et VCaP cells by GSH, inhibitors of TGFβ (S) and BMP (K) receptors, or their combinations (KS and GSK). Cloning efficiency were measured in non-treated cells (Ø) or in cells treated for 7 d with 0.2 nM R1881 followed by a culture for additional 20 d in growth medium with the indicated compounds. Values are the mean ± sd of 2 independent experiments. Asterisks indicate the statistical significance of the difference in cloning efficiency between cells treated only with R1881 and cells under other culture conditions.
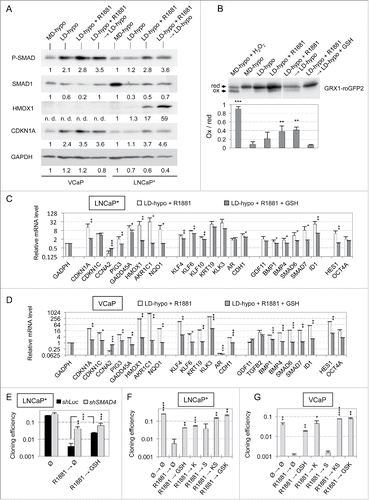
To assess redox imbalance, we used a cellular thiol-specific redox sensor (GRX1-roGFP2) formed by the redox-sensitive green fluorescent proteins (roGFP2) fused to the GRX1 protein, that allows rapid equilibration of the roGFP2 moiety with the cell glutathione/glutathione disulfide (GSH/GSSG) redox couple.Citation25 Thus, the ratio of oxidized to reduced roGFP2 measured in the cell can be used to generate an in vivo readout of the GSH/GSSG redox state. The oxidized and reduced forms of roGFP2 can be easily separated by nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), providing a sensitive and highly reproducible fluorescence-independent approach of redox analysis. Accordingly, LNCaP* cells were stably transduced with a retroviral vector expressing GRX1-roGFP2 and the relative levels of reduced and oxidized GRX1-roGFP2 were measured in 3 independent cell populations by redox Western blot analysis. As shown in and S5, the level of oxidized GRX1-roGFP2 was barely detectable in cells cultured at medium cell density but increased about twofold in cells cultured at low cell density for 7 d. Although this effect did not reach statistical significance, it was in agreement with our previous finding that the mere culture at low cell density was sufficient to increase expression of stress markers (Citation2 and ). The relative level of oxidized GRX1-roGFP2 further increased significantly after a 7 day-treatment with R1881 ( and S5). A high level of oxidized GRX1-roGFP2 was maintained after withdrawal of R1881, indicating that the more oxidized glutathione redox state has become self-sustained in dormant cells ( and S5). This oxidative shift of glutathione redox potential was fully reversed when glutathione was added in the growth medium after withdrawal of R1881 at the 7th day, consistent with the antioxidant effect of glutathione ( and S5). Several additional observations supported that cells sensed these redox changes. First, the oxidative shift of glutathione redox potential in LNCaP* cells correlated with an increased expression of the oxidative stress markers CDKN1A and HMOX1, expression of HMOX1 becoming maximal when roGFP2 sensor was maximally oxidized after withdrawal of R1881 ( and S5). Second, RT-qPCR analysis of LNCaP* or VCaP cells showed that GSH, when added with androgens, suppressed the induction of nearly all dormancy signature genes, including all markers of cellular stress (). These changes in genes expression resulting from GSH addition in androgen-treated cells were inversely related to those induced by a pro-oxidant treatment with hydrogen peroxide in cells cultured at medium cell density (Pearson correlation coefficient r > 0.92; p<0.0001, Table S1), strongly suggesting that effects of GSH in androgen-treated cells were fully accounted by inhibition of a pre-existing oxidative stress. Finally, the functional involvement of redox imbalance in androgen-induced dormancy was supported by the fact that thiol antioxidants such as GSH and NAC were able to partially revert androgen-induced dormancy when added after androgen withdrawal ( and S4).
Androgen treatment of LNCaP* cells at medium cell density induced an unstable quiescent state correlated with a diminished induction of oxidative stress
To define the role of cell density in androgen-induced dormancy, we analyzed the effects of androgen treatment on cell cultured at medium cell density. R1881 at 0.2 nM had no significant effect on the rate of VCaP cell proliferation () whereas it progressively decreased proliferation rate of LNCaP*, a complete growth arrest being observed after an exposure to R1881 for 11 d (). However, this growth arrest was unstable as cells resumed proliferation 4 d after withdrawal of R1881 (). Long-term treatment with 0.5 nM DHT (12 days) or shorter duration treatment with 0.2 nM R1881 (7 days) induced weaker effects, failing to reach a complete cell growth arrest and quicker recovery after washing out of the exogenous androgens (Fig. S6 and data not shown). To get more insights into the differential effects of androgens in relation to cell density, we compared the effects of R1881 on expression of the dormancy signature genes for cells cultured at low and medium density. VCaP cells at medium cell density displayed no significant change regarding the expression of these genes (except for KLK3) following 11 d in the presence of R1881, suggesting that neither TGFβ/BMP nor redox imbalance signaling was activated under these conditions (). In contrast, LNCaP* cells at medium cell density displayed a pattern of gene expression after 11 d in the presence of R1881 which was similar to that found in cells treated at low cell density for 7 d. Notably, however, a decreased induction of oxidative stress markers like AKR1C1, HMOX1 and NQO1 was observed, leading to a Pearson correlation coefficient of 0.65 (). A more complete concordance (r = 0.89) could be observed if the pattern of gene expression induced by R1881 at medium density was compared with that of cells at low density in the presence of both R1881 and GSH (). Western blot analysis confirmed that R1881 treatment of cells at medium cell density failed to induce HMOX1 expression, as well as CDKN1A in spite of an increased mRNA level ( and S5). These observations suggested that redox imbalance induced by R1881 at medium cell density was weaker than that induced at low cell density. Consistently, measurement of oxidization state of the GRX1-roGFP2 sensor confirmed that there was at most a slight redox imbalance under such consition (), even less than that induced by the mere cell culture at low cell density, in contrast to the significantly increased oxidation of sensor observed in . We also noted that R1881 induced an about twofold increase of SMAD phosphorylation in LNCaP* cells (), which correlated with induction of markers of SMAD activation like SMAD7 and ID1 (), indicating that TGFβ/BMP signaling was activated by R1881 at medium cell density. Addition of the K02288, an inhibitor of BMP receptors, or of glutathione alleviated the growth-inhibitory effect of R1881 on cells at medium density (). This suggested that mechanisms involved in the growth-arrest induced by R1881 at medium cell density were in part similar to those involved in dormancy but that the instability of the growth-arrest correlated with a diminished induction of oxidative stress and oxidative stress markers like CDKN1A.
Figure 5. Blunted induction of dormancy by androgens in cells cultured at medium cell density. (A and B) Cumulated cell population doublings of VCaP and LNCaP* cells, respectively, grown under MD-hypo condition in the presence of none or R1881 as indicated. Data are derived from 2 independent experiments. (C and D) Differential effects of R1881 (0.2 nM) on the expression of the dormancy signature genes depending on density of cultured VCaP and LNCaP* cells, respectively. Values are the mean ± sd of 2 independent RT-qPCR experiments with value measured under MD-hypo condition being set at 1. Asterisks indicated statistical significance for the differences in mRNA levels between cells cultured at low (LD-hypo + R1881 for 7 days) and medium (MD-hypo + R1881 for 11 days) density. (E) Pearson correlation coefficients between the expression profile of the dormancy signature genes induced by a R1881 treatment of LNCaP* cultured at medium cell density for 11 d (MD-hypo + 0.2 nM R1881) and those induced by other indicated cell culture conditions. Values are from data used in Fig. S1, 4C and 5D. (F) Western blot analysis of SMAD phosphorylation and SMAD1 and CDKN1A expression in LNCaP* and VCaP cells according to the indicated culture conditions. Numbers indicate the relative levels of GADPH or the relative amount of the indicated proteins normalized by GAPDH levels. (G) Oxidation level of the GRX1-roGFP2 sensor in LNCaP* cells treated by 0.2 nM of R1881 for 11 d at medium cell density. The lower panel indicates the ox/red ratio of GRX1-roGFP2 under the indicated cell culture conditions. Values (mean ± sd) were derived from 3 independently transduced cell populations. (H) Cumulated cell population doubling of LNCaP* cells grown at medium cell density (MD-hypo) in the presence of none, 0.2 nM R1881, 0.2 µM K02288 (K), 0.2 µM SB505124 (S) and 8 mM glutathione (G) or their combination as indicated. Data are averaged from 2 independent experiments.
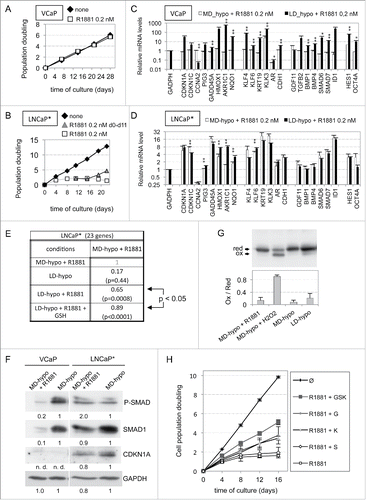
Transient CDKN1A overexpression in cells cultured at low cell density is sufficient to induce a self-sustained quiescent state regulated by redox imbalance and SMAD signaling
To better characterize the cell signaling pathways involved in redox imbalance in dormant cells, we focused on the role of CDKN1A protein (p21CIP1) since its level correlated tightly with the degree of redox imbalance (). A functional involvement of CDKN1A in androgen-induced dormancy was investigated by examining the effects of its depletion. Targeting of CDKN1A with a doxycyline (dox)-inducible specific shRNA (shp2126) induced ∼75% decrease of both CDKN1A mRNA and protein (p21CIP1/waf1/Sdi1) levels (). Remarkably, compared with cells transduced with a control vector, CDKN1A depletion increased by about 7 times the cloning efficiency of cells treated with R1881 for 7 d (). However, effect of CDKN1A depletion was diminished when it was started after the 7-days-treatment with R1881 (). This suggested that CDKN1A played a significant role in the establishment of the dormant state. To further investigate this point, we examined whether transient overexpression of CDKN1A in cells cultured at low cell density could substitute for androgen or hypertonicity to induce a self-sustained growth arrest. Accordingly, we derived LNCaP* cell populations that were transduced with a dox-inducible control (pTRIP-control) or CDKN1A expressing (pTRIP-p21) lentiviral vector. We observed that induction of pTRIP-p21 expression by dox- for 7 d in cells cultured at low density was sufficient to decrease their cloning efficiency by ∼100 fold as compared with control cells (). This inhibitory effect was partially reversed when glutathione or pharmacological inhibitors of BMP and TGFβ receptors were added after the washing-out of dox at the 7th day and nearly fully reversed when both were added simultaneously (). Western blot analysis showed that levels of expression of HMOX1 and CDKN1A were increased ∼2 and ∼4 times respectively in pTRIP-p21 transduced cells after a 7-day dox treatment, and these levels were further augmented 1.5 and 2 times respectively at day 14, 7 d after withdrawal of dox (). This implied that endogenous CDKN1A has been induced, since RT-qPCR analysis showed that expression of CDKN1A transcripts derived from the transduced lentiviral vector sharply decreased to nearly background levels 7 d after dox withdrawal at day 14 (). SMAD phosphorylation also increased about 2-fold by day 14, but no significant increase could be detected at day 7 at the end of dox treatment (). Of note, dox displayed no such effect in control cells (), suggesting that transient exogenous CDKN1A overexpression for 7 d at low cell density was sufficient to trigger endogenous CDKN1A and HMOX1 expression and SMAD phosphorylation in a self-sustained manner. These observations were confirmed by RT-qPCR analysis of the expression of the dormancy signature genes. Transient CDKN1A overexpression for 7 d induced some changes in the level of expression for most of the dormancy signature genes, but these changes were further amplified after an additional 7-day culture in dox-free medium (). Indeed, as ascertained by Pearson's correlation coefficients (), the pattern of gene expression induced by CDKN1A displayed a high level of similarity with that of dormant LNCaP* cells induced by transient exposure to androgens only at day 14 (). To test whether this dormant state reached at day 14 used the same self-sustaining mechanisms as the dormant states induced by the androgens or exposure to a hypertonic medium, we exposed cells to glutathione and inhibitors of TGFβ/BMP receptors. As shown in , glutathione or inhibitors of TGFβ/BMP receptors could almost fully reverse this growth arrest when added in combination, a partial reversion being observed when these compounds were used separately. In contrast to these long-term effects observed in cells at low density, CDKN1A overexpression in cells at medium density induced only a transient growth-arrest, cells resuming proliferation immediately after withdrawal of dox (Fig. S6). Taken together, our data showed that the induction of dormancy by transient CDKN1A overexpression, exposure to androgens or to hypertonicity shared not only the same self-sustaining mechanisms but also a marked dependency on cell density.
Figure 6. Regulation of androgen-induced dormancy by CDKN1A depletion. (A) Scheme of the experimental design for CDKN1A depletion. Cells were cultured under LD-hypo conditions in the presence of doxycycline (dox) and/or 0.2 nM R1881 for 7 days, washed, and then further cultured for additional 20 d in the presence of dox. (B) Western blot analysis of CDKN1A expression in control and pTRIP-shp21 transduced LNCaP* cells cultured under LD-hypo + dox for 7 d. (C) Relieving of androgen-mediated inhibition of cell cloning by early depletion of CDKN1A. Values are the mean ± sd derived from 2 independently transduced cell populations with control and pTRIP-shp21 lentiviral vectors. Statistical significance of the cloning efficiency difference between control and pTRIP-shp21 transduced LNCaP* cells is indicated (black asterisks).
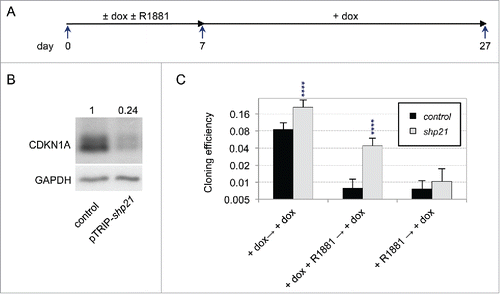
Figure 7. Induction of a dormant state related to androgen-induced dormancy through transient overexpression of CDKN1A. (A) Scheme of the experimental design for transient CDKN1A overexpression. Transduced cell populations were cultured under LD-hypo conditions in the presence of none or doxycycline (dox) to induce CDKN1A expression for 7 d and then cultured in the presence of GSH and/or inhibitors of TGFβ/BMP receptors as described in (LD-hypo + dox → LD-hypo ± none (Ø) or GSH or KS or GSK). RT-qPCR and Western blot analysis were performed at day 7 (LD-hypo, LD-hypo + dox) or at day 14 (LD-hypo + dox → LD-hypo). (B) Cloning efficiencies of control and pTRIP-p21-transduced LNCaP* cells following transient dox treatment and subsequent reversal in the presence of GSH, inhibitors of TGFβ/BMP receptors (KS) or combination of GSH and inhibitors of TGFβ/BMP receptors (GSK). Statistical significance of the difference in cloning efficiency between control and pTRIP-p21 transduced cells treated only by dox (large asterisk) and of the effects of GSH, KS and GSK (compared with the same cell populations not treated by these compounds; small asterisks) are indicated. (C) Western blot analysis of SMAD phosphorylation or SMAD1, HMOX1 and CDKN1A expression. Cells were cultured as indicated. Numbers indicate the relative levels of GADPH or the relative amount of the indicated proteins normalized by GAPDH levels. Similar data were obtained with another independent couple of control and pTRIP-p21 transduced cell populations. (D) RT-qPCR analysis of exogenous p21 mRNA level transcribed from transduced pTRIP-p21 vector under the indicated cell culture conditions. The couple of primers targeted the 5’ part of the UbqC promoter just downstream of CDKN1A ORF in the pTRIP-p21 expression vector. Control is non-transduced LNCaP* cells. Data were normalized as in . Variations between any couple of different cell culture conditions were significant (p < 0.05). (E) RT-qPCR analysis of the expression of the dormancy signatures genes induced by transient overexpression of CDKN1A. mRNA levels were normalized as in ). Asterisks indicated statistical significance for the differences in mRNA levels between control cell populations cultured under LD-hypo and pTRIP-p21-transduced cells cultured for 7 d in the presence of dox followed by an additional 7 d under LD-hypo conditions (LD-hypo + dox → LD-hypo). (F) Comparison between the expression profiles of dormancy signature genes from dormant cells induced by exposure to R1881 or overexpression of CDKN1A. Data are derived from those used in . (G) Reversal of CDKN1A overexpression-induced dormancy through a delayed treatment with GSH (G) or inhibitors of BMP and TGFβ receptors (KS). These compounds were added only at day 14 and cells were thereafter cultured for additional 18 d for cloning efficiency measurements. Statistical significance of the cloning efficiency difference relative to the cells treated with dox alone is indicated. All data from this figure are derived from experiments with 2 independently transduced cell populations with control and pTRIP-p21 lentiviral vectors.
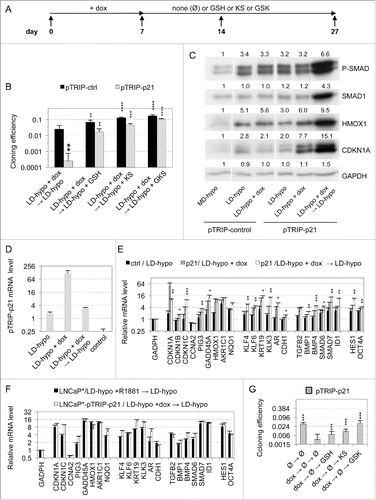
Table 1. SimilarityFootnote1 in genes expression profiles between dormant cells induced by transient exposure to R1881 and by transient CDKN1A overexpression at low cell density.
Discussion
In this work, we characterized a self-sustained quiescent state that could be induced in androgen-sensitive prostate cancer cells cultured at low density by exposure to androgens or ectopic expression of CDKN1A. The maintenance of this quiescent state no longer required the inducer as we have previously suggested for the quiescence induced by a slightly hypertonic mediumCitation1 and was dependent upon a redox imbalance and a TGFβ/SMD signaling. Thus, several inducers targeting different biologic pathways could lead to a similar self-sustained quiescent state. Moreover, reducing redox imbalance or TGFβ/BMP signaling allowed cells to resume growth indicating that, although stable and self-sustained, this quiescence could be reversed and qualified as a dormant state.
In each of these dormancy phenomena, we could distinguish an induction phase from a maintenance phase (Fig. S7). Thus, while AR antagonists could block entry in quiescence when added at the same time as the androgen, they were no longer effective when added later (compare ). Similarly, switching cells from hypertonic to hypotonic conditions was not sufficient to reverse quiescence once it had been established.Citation1 Induction of dormancy as well as establishment of the maintenance phase took place over a time scale of several days. Indeed, although cells at low cell density displayed the essential features of dormancy after a 7-day treatment with androgens (), the dormant state was not yet fully established since SMAD phosphorylation and CDKN1A protein level (as well as of HMOX1 for LNCaP* cells) further augmented 7 d later (). Of note, transient overexpression of CDKN1A was markedly slower in inducing the expression profile of dormancy (compare correlation coefficient at day 7 and 14 in ). Thus, SMAD phosphorylation was observed only after 14 d while the levels of CDKN1A and HMOX1 were still increasing. These observations support a model in which inducers of dormancy set up a progressive reprogramming of cells leading to a growth arrest which then self-stabilizes itself by positive feedback loops. In agreement, changes in genes expression occurring over a long period have been documented in others types of cell quiescence,Citation27 suggesting that most quiescent states require an extensive reprogramming of gene expression.
Whether dormancy is induced by androgens, transient expression of CDKN1A (and likely by a slightly hypertonic medium), the maintenance phase is dependent upon an activation of TGFβ/BMP signaling and a redox imbalance (Fig. S7). Importantly, although these 2 pathways induce distinct, albeit pleiotropic responses, they are strongly interconnected in dormant cells since suppression of redox imbalance with reduced thiols strongly inhibited the expression of TGFβ/BMP-SMAD signaling targets ( and see inCitation2), and reciprocally, the induction of oxidative stress markers such as HMOX1 and AKRC1 was reduced upon inhibition of SMAD signaling (Fig. S3 and see Table S4 inCitation2). An interconnection between redox imbalance and SMAD signaling has been reported in other cellular contexts (see for instanceCitation28-34), suggesting that it has broad biologic implications. Importantly, this could in part explain how distinct inducing signaling pathways converge onto similar end-states (Fig. S7). That the processes initially activated by androgens and hypertonicity differ was demonstrated by the ineffectiveness of AR antagonists to prevent dormancy induced by hypertonicity (data not shown). The observation that CDKN1A silencing could prevent the establishment of dormancy by androgens establishes that CDKN1A is part of the cellular response to androgens. It should not be taken, however, as indicator that androgens and CDKN1A act through the same pathways. Indeed, beyond the difference in kinetics discussed above, CDKN1A overexpression, in contrast to androgens, primarily enhanced redox imbalance since it enhanced expression of the oxidative stress marker HMOX1 before activating SMAD phosphorylation (). Rather, these observations are in agreement with a slow convergence of different reprogramming cascades enabled by the coupling of the 2 central effectors which are redox imbalance and TGFβ/BMP signaling.
Thus, as for senescence, multiple inducers can induce closely similar cellular states indicating that the dormant state constitutes an important attractor in the phenotypic space. However this is valid only for cells cultured at low cell density since these inducers failed to efficiently activate these signaling pathways in cells at medium density (this work, data not shown andCitation2). Interestingly, the synthetic androgen R1881 could induce a growth arrest in LNCaP* cells cultured at medium cell density which was dependent upon an induction of TFGβ/BMP and redox signaling. However, this growth-arrest was unstable as it persisted for only a few days after androgen withdrawal, which correlated with a lack of induction of some oxidative stress markers like CDKN1A. Thus, our data indicates that, at medium density, cells are not completely refractory to dormancy inducers but their partial response fails to induce a self-sustaining oxidative stress and TGFβ/BMP signaling. This suggests that homotypic cellular interactions can strongly impact the primary response to dormancy inducers and/or the establishment of the self-sustaining loops enabling a stable quiescence. In contrast, heterotypic interactions with non-cancerous cells could promote cancer cell quiescence as suggested by 3D co-culture experiments of breast cancer cells with mixed cell typesCitation35 and in vivo observations showing that disseminated melanoma and breast cancer cells are highly prone to enter into a dormant state after their extravasation into the parenchyma of distant organs.Citation36-40 Accordingly, when LuCaP prostate cancer cells were incubated on a monolayer of bone marrow stromal cells, active proliferation could only be observed when cells were seeded at high density.Citation41 Similarly, in a model of quiescence of breast cancer cells induced by culture in Matrigel, cells could escape this quiescence when their density exceeded a threshold allowing homotypic interactions.Citation42 Taken together, these observations suggest that entry into dormancy and the establishment of a self-sustained quiescence require critical thresholds of TGFβ/BMP signaling and redox imbalance which can only be achieved in the absence of homotypic interactions.
Our findings presented in this study may have potential therapeutic applications. It is known that more than 10% of patients treated with radical prostatectomy for prostate cancer will have a metastatic relapse of their cancer within the next 10 years, most likely due to an early metastatic spread of cancer cells that colonizes distant organs as solitary cancer cells.Citation43 Therefore, transient treatments with androgens could enhance their transition toward a dormant state and/or stabilize their dormancy, thereby delaying the development of clinical metastases. This would be effective if the treatment is started just after radical prostatectomy, at a time when cancer cells are still dispersed and solitary, since, as indicated by our data, clusters of interacting cancer cells may become resistant to induction of dormancy. This therapy should be complementary to medical castration as androgen deprivation is likely to be a driving force which leads prostate cancer cells to become hypersensitive to androgens and to enter into dormancy in response to physiologic levels of androgens. Moreover, repeated cycles of androgen deprivation and supplementation could be envisioned, as AR antagonists does not destabilize dormancy once established (). It is noteworthy that a similar paradigm of bipolar androgen therapy (BAT) was previously proposed to counteract the adaptative autoregulation of AR expression to the change of androgen concentration in CRPC cells.Citation14,21 Our data suggest that BAT could also be of therapeutic value if applied earlier before biochemical relapse and at more distant intervals to inhibit proliferation of androgen-dependent disseminated cancer cells when they are still solitary. Thus, even if this treatment does not provide a complete protection from relapse it could be a welcomed therapeutic option since a reduction in the frequency of escape from dormancy should translate into a proportional extension of progression-free survival at a very limited cost.
Materials and methods
Plasmids, chemicals and primary antibodies
Plasmid pTRIPz-shp21 (Clone Id: V2THS_202469, ref. RHS4696–200673680 from Open Biosystems) and pBabepuro-shSMAD4 were previously characterized.Citation2,26,44 Doxycycline (dox)-inducible pTRIP-p21 vector was constructed by inserting a blunt-ended BamHI-EcoRI human CDKN1A cDNA fragment excised from p21-pBabe-puro (a kind gift from Gordon Peters, Cancer Research UK – London Research Institute) in place of the RFP-shRNA cassette of pTRIPz-shp21 excised upon AgeI to MluI restriction followed by klenow treatment. pFbneo-GRX1-roGFP2 vector was constructed by ligating the SmaI-NotI-digested GRX1-roGFP2 DNA fragment excised from plasmid pEIGW-GRX1-roGFP2Citation25 (a kind gift from Tobias Dick, Addgene plasmid # 64990) to the EcoRI-cut and klenow-treated and Not1-cut pFbneo vector (Stratagene). K02288 was from Tocris (ref. 4986), SB505124 was from Abcam (ref. ab144402), reduced L-glutathione (ref. G6013) and N-ethylmaleimide (ref. E3876) from Sigma-Aldrich. Primary antibodies used were anti-CDKN1A (ref. sc-6246, Santa Cruz Biotechnology), anti-HMOX1 (ref. sc-136960, Santa Cruz Biotechnology), anti-Phospho-SMAD1/5 (Ser463/465) (ref. 9516, Cell Signaling Technology), anti-SMAD1 (ref. 6944, Cell Signaling Technology), anti-GAPDH (ref. 5174, Cell Signaling Technology) and anti-GFP (ref. A-11122, ThermoFisher Scientific).
Cell culture, recombinant retrovirus production, infection and flow cytometry
All cells were cultured at 37°C in 5% CO2 atmosphere. Early passage LNCaP and 22Rv1 cells were a kind gift from A. Chauchereau (Gustave Roussy Institute, France) and were maintained in isotonic RPMI-1640 medium supplemented with stabilized glutamine (ref. 61870044, GIBCO, ThermoFisher Scientific), 10% fetal calf serum (FCS, PAA Laboratories) and penicillin plus streptomycin 1X solution (GIBCO, ThermoFisher Scientific). LNCaP* and VCaP prostate cancer cells were cultured in a hypotonic growth medium (hypo) made by supplementation of DMEM-FCS with 25% H2O, which was previously shown to be optimal for cloning of LNCaP* and VCaP cells (Citation2 and data not shown). DMEM-FCS referred to DMEM (containing 862 mg/l glutamax-I, 4.5 g/l glucose and 1 mM pyruvate, ref. 31966, GIBCO, ThermoFisher Scientific) supplemented with 10% FCS (PAA Laboratories) and penicillin plus streptomycin 1X solution. pH of DMEM-FCS + 25% H2O under a 5% CO2 atmosphere was 7.34 ± 0.05 (mean ± sd derived from 5 independent measurements performed after a 1, 3 and 10 day equilibration period in the cell incubator. MD-hypo and LD-hypo specifically referred to cell culture in hypotonic DMEM-FCS at medium and low cell density, respectively. Medium cell density referred to 2 × 104 seeded cells cm−2while low cell density corresponded to 25 seeded cells cm−2 for 22Rv1, 50 seeded cells cm−2 for native LNCaP and 102 seeded cells cm−2 for LNCaP*. For cloning efficiency experiments, cells were cultured as indicated at low cell density and the number of colonies per dish counted directly by microscope examination (for VCaP cells) or after fixing in 10% glacial acetic acid plus 20% ethanol and staining with a solution containing 0.2% crystal violet (ref. C6158 Sigma-Aldrich) in 10% glacial acetic acid and 20% methanol (for the other cell lines and a few experiments with VCaP). Cloning efficiency was calculated as the ratio of the number of cell colonies per dish to the number of seeded cells per dish. Recombinant retroviruses were produced as described previously, PVPack-gagpol plasmid (Stratagene) and pCMV-DR8.74 being used for lentiviral and murine recombinant retroviruses, respectively.Citation2 Viral transduction and flow cytometry for analysis of cell DNA content were performed as previously.Citation1 Induction of lentiviral vectors expression was performed with doxycycline (dox) at 0.5 µg/ml. All primary stocks of transduced cell populations were frozen in multiple aliquots and tested for mycoplasma contamination about one month after thawing with a home-made RT-qPCR detection kit including positive controls dissolved in culture medium.
Western blot analysis under non-reducing conditions
Cells were incubated with 20 mM N-ethylmaleimide (NEM) in PBS (4 ml for a 10-cm diameter dish) for 5 min at room temperature, harvested by pipetting after addition of FCS to 0.5% and EDTA to 1 mM final concentrations, washed with cold PBS and lysed by a 30 min incubation on ice in EBC lysis buffer (50 mM Tris-HCl pH 8, 120 mM NaCl, 10 mM EDTA and EGTA, Protease inhibitors cocktail (ref. P8340, Sigma-Aldrich) diluted 100 times, 1 mM PMSF, 20 mM NaF and 10 mM sodium orthovanadate). The supernatant recovered after a 10-min centrifugation at 13,000 rpm and 4°C was aliquoted for protein quantitation according to a modified Bradford protocolCitation2 or mixed with Laemmli buffer without β-mercaptoethanol and boiled for 5 min before loading for SDS-PAGE electrophoresis. Proteins were blotted electrophoretically onto a 0.45-µm-pore-size polyvinylidene difluoride Hybond P membrane (ref. 10600023, Amersham). Immunodetection was performed by using a Horseradish Peroxidase detection kit (ECL Select detection kit, Amersham) with the indicated primary antibody and high performance chemiluminescence film (Hyperfilm ECL, Amersham). Films were digitalized into TIFF format with a trans-illuminating scanner (Scan Epson Perfection 4490 Photo, 16 bits gray at 1200 dpi) and relative intensity of close bands were measured with the Image Studio Lite Western Blot analysis software (LI-COR Biotechnology)
Western blot analysis under reducing conditions
It was performed essentially as above except that NEM treatment was omitted and denaturation of proteins was performed in complete Laemmli buffer with 100 mM β-mercaptoethanol. Additionally, quantification of the relative intensity of well-separated bands was performed with the ImageJ software following the standard procedure for gel quantification.
RT-qPCR assays
RT-qPCR analysis was performed as described in detail in.Citation2 In these assays, GAPDH was used as a reference gene for normalization of data derived from independent cDNA synthesis reactions since it showed only small variations in its level of expression between different cell culture conditions (on average, less than 50% variation as normalized to the amount of total RNA added into the cDNA synthesis reaction). The levels of each mRNA species were also normalized to that measured under MD-hypo condition, set at 1 (arbitrary unit) in all experiments. Primers sequences hybridizing to the Ubqc promoter in pTRIP-p21 were Ubqc1-F: AGACTCGGCCTTAGAACC and Ubqc1-R: AGACTCGGCCTTAGAACC. Others primers sequences were given inCitation2
Statistical analysis
Data are presented as mean ± standard deviation (mean ± sd). Significance (p value) of variations was calculated using a paired or a heteroscedatic Student's t test with unilateral distribution and was indicated as **** for p < 0.002, *** for 0.002 < p < 0.01, ** for 0.01 < p < 0.05 and * for 0.05 < p < 0.1. For correlation analysis between patterns of mRNA accumulation measured under different cell culture conditions, the linear correlation coefficient of Pearson was calculated for the dormancy signature genes as previously.Citation2 Significance of the correlation coefficients was calculated on the VassarStats website (http://vassarstats.net/rsig.html).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
Conceptualization and Supervision: TT; Methodology: MEH and TT; Investigation: ATB, FLT, MEH, MH, and TT; Analysis and Visualization of Data: ATB, FLT and TT; Writing original draft: TT; Writing-Review and Editing: ATB, FD, MEH, TT; Funding acquisition: MEH and TT.
1310345_Supplemental_Material.zip
Download Zip (657.2 KB)Acknowledgments
T. T. is grateful to Peters Gordon and Tobias Dick for providing plasmids and to Anne Chauchereau for the gift of native LNCaP and 22Rv1 cells, and of R1881 and Enzalutamide.
Funding
This work was supported by the Center National de la Recherche Scientifique (CNRS). M-E.H. is also supported by the Institut Curie. A.T.B. is a recipient of a doctoral fellowship from the ‘Ecole Doctorale de Cancérologie de Paris-Saclay’.
Related Research Data
References
- Havard M, Dautry F, Tchenio T. A dormant state modulated by osmotic pressure controls clonogenicity of prostate cancer cells. J Biol Chem 2011; 286:44177-86; PMID:22039055; http://dx.doi.org/10.1074/jbc.M111.262709
- Bui AT, Laurent F, Havard M, Dautry F, Tchenio T. SMAD signaling and redox imbalance cooperate to induce prostate cancer cell dormancy. Cell Cycle 2015; 14:1218-31; PMID:25706341; http://dx.doi.org/10.1080/15384101.2015.1014145
- Wang ZA, Shen MM. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene 2011; 30:1261-71; PMID:21119602; http://dx.doi.org/10.1038/onc.2010.530
- Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell 2012; 21:253-65; PMID:22340597; http://dx.doi.org/10.1016/j.ccr.2012.01.005
- Wadosky KM, Koochekpour S. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget 2016; 7:64447-70; PMID:27487144; http://dx.doi.org/10.18632/oncotarget.10901
- Langeler EG, van Uffelen CJ, Blankenstein MA, van Steenbrugge GJ, Mulder E. Effect of culture conditions on androgen sensitivity of the human prostatic cancer cell line LNCaP. Prostate 1993; 23:213-23; PMID:7694266; http://dx.doi.org/10.1002/pros.2990230304
- Umekita Y, Hiipakka RA, Kokontis JM, Liao S. Human prostate tumor growth in athymic mice: Inhibition by androgens and stimulation by finasteride. Proc Natl Acad Sci U S A 1996; 93:11802-7; PMID:8876218; http://dx.doi.org/10.1073/pnas.93.21.11802
- Kokontis JM, Hay N, Liao S. Progression of LNCaP prostate tumor cells during androgen deprivation: Hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol Endocrinol 1998; 12:941-53; PMID:9658399; http://dx.doi.org/10.1210/mend.12.7.0136
- Tsihlias J, Zhang W, Bhattacharya N, Flanagan M, Klotz L, Slingerland J. Involvement of p27Kip1 in G1 arrest by high dose 5 alpha-dihydrotestosterone in LNCaP human prostate cancer cells. Oncogene 2000; 19:670-9; PMID:10698512; http://dx.doi.org/10.1038/sj.onc.1203369
- Chuu CP, Hiipakka RA, Fukuchi J, Kokontis JM, Liao S. Androgen causes growth suppression and reversion of androgen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res 2005; 65:2082-4; PMID:15781616; http://dx.doi.org/10.1158/0008-5472.CAN-04-3992
- Tararova ND, Narizhneva N, Krivokrisenko V, Gudkov AV, Gurova KV. Prostate cancer cells tolerate a narrow range of androgen receptor expression and activity. Prostate 2007; 67:1801-15; PMID:17935158; http://dx.doi.org/10.1002/pros.20662
- Vander Griend DJ, Litvinov IV, Isaacs JT. Stabilizing androgen receptor in mitosis inhibits prostate cancer proliferation. Cell Cycle 2007; 6:647-51; PMID:17387277; http://dx.doi.org/10.4161/cc.6.6.4028
- Chuu CP, Kokontis JM, Hiipakka RA, Fukuchi J, Lin HP, Lin CY, Huo C, Su LC, Liao S. Androgen suppresses proliferation of castration-resistant LNCaP 104-R2 prostate cancer cells through androgen receptor, Skp2, and c-Myc. Cancer Sci 2011; 102:2022-8; PMID:21781227; http://dx.doi.org/10.1111/j.1349-7006.2011.02043.x
- Isaacs JT, D'Antonio JM, Chen S, Antony L, Dalrymple SP, Ndikuyeze GH, Luo J, Denmeade SR. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate 2012; 72:1491-505; PMID:22396319; http://dx.doi.org/10.1002/pros.22504
- Mirochnik Y, Veliceasa D, Williams L, Maxwell K, Yemelyanov A, Budunova I, Volpert OV. Androgen receptor drives cellular senescence. PloS One 2012; 7:e31052; PMID:22403609; http://dx.doi.org/10.1371/journal.pone.0031052
- Thelen P, Heinrich E, Bremmer F, Trojan L, Strauss A. Testosterone boosts for treatment of castration resistant prostate cancer: An experimental implementation of intermittent androgen deprivation. Prostate 2013; 73:1699-709; PMID:23868789; http://dx.doi.org/10.1002/pros.22711
- Roediger J, Hessenkemper W, Bartsch S, Manvelyan M, Huettner SS, Liehr T, Esmaeili M, Foller S, Petersen I, Grimm MO, et al. Supraphysiological androgen levels induce cellular senescence in human prostate cancer cells through the Src-Akt pathway. Mol Cancer 2014; 13:214; PMID:25216853; http://dx.doi.org/10.1186/1476-4598-13-214
- Sedelaar JP, Isaacs JT. Tissue culture media supplemented with 10% fetal calf serum contains a castrate level of testosterone. Prostate 2009; 69:1724-9; PMID:19676093; http://dx.doi.org/10.1002/pros.21028
- Makkonen H, Kauhanen M, Jaaskelainen T, Palvimo JJ. Androgen receptor amplification is reflected in the transcriptional responses of vertebral-cancer of the prostate cells. Mol Cell Endocrinol 2011; 331:57-65; PMID:20728506; http://dx.doi.org/10.1016/j.mce.2010.08.008
- Marcias G, Erdmann E, Lapouge Gl, Siebert C, Barthélémy P, Duclos B, Bergerat J-P, Céraline J, Kurtz J-E. Identification of novel truncated androgen receptor (AR) mutants including unreported pre-mRNA splicing variants in the 22Rv1 hormone-refractory prostate cancer (PCa) cell line. Hum Mutat 2010; 31:74-80; PMID:19830810; http://dx.doi.org/10.1002/humu.21138
- Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, Luo J, Haffner MC, Yegnasubramanian S, Carducci MA, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: Results from a pilot clinical study. Sci Transl Med 2015; 7:269ra2-ra2; PMID:25568070; http://dx.doi.org/10.1126/scitranslmed.3010563
- Olsen JR, Azeem W, Hellem MR, Marvyin K, Hua Y, Qu Y, Li L, Lin B, Ke XIS, Øyan AM, et al. Context dependent regulatory patterns of the androgen receptor and androgen receptor target genes. BMC Cancer 2016; 16:377; PMID:27378372; http://dx.doi.org/10.1186/s12885-016-2453-4
- Sanvitale CE, Kerr G, Chaikuad A, Ramel MC, Mohedas AH, Reichert S, Wang Y, Triffitt JT, Cuny GD, Yu PB, et al. A new class of small molecule inhibitor of BMP signaling. PloS One 2013; 8:e62721; PMID:23646137; http://dx.doi.org/10.1371/journal.pone.0062721
- Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFbs and BMP pathways. Cellular Signal 2011; 23:1831-42; PMID:21740966; http://dx.doi.org/10.1016/j.cellsig.2011.06.019
- Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods 2008; 5:553-9; PMID:18469822; http://dx.doi.org/10.1038/nmeth.1212
- Zemskova M, Lilly MB, Lin YW, Song JH, Kraft AS. p53-dependent induction of prostate cancer cell senescence by the PIM1 protein kinase. Mol Cancer Res 2010; 8:1126-41; PMID:20647331; http://dx.doi.org/10.1158/1541-7786.MCR-10-0174
- Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol 2006; 4:e83; PMID:16509772; http://dx.doi.org/10.1371/journal.pbio.0040083
- Li H, Sekine M, Seng S, Avraham S, Avraham HK. BRCA1 interacts with Smad3 and regulates Smad3-mediated TGF-beta signaling during oxidative stress responses. PloS One 2009; 4:e7091; PMID:19768112; http://dx.doi.org/10.1371/journal.pone.0007091
- Zhu D, Wu J, Spee C, Ryan SJ, Hinton DR. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J Biol Chem 2009; 284:9529-39; PMID:19158083; http://dx.doi.org/10.1074/jbc.M809393200
- Dyer LA, Pi X, Patterson C. The role of BMPs in endothelial cell function and dysfunction. Trends Endocrinol Metab 2014; 25:472-80; PMID:24908616; http://dx.doi.org/10.1016/j.tem.2014.05.003
- Kretova M, Sabova L, Hodny Z, Bartek J, Kollarovic G, Nelson BD, Hubackova S, Luciakova K. TGF-beta/NF1/Smad4-mediated suppression of ANT2 contributes to oxidative stress in cellular senescence. Cellular Signal 2014; 26:2903-11; PMID:25220407; http://dx.doi.org/10.1016/j.cellsig.2014.08.029
- Hubackova S, Kucerova A, Michlits G, Kyjacova L, Reinis M, Korolov O, Bartek J, Hodny Z. IFNgamma induces oxidative stress, DNA damage and tumor cell senescence via TGFbeta/SMAD signaling-dependent induction of Nox4 and suppression of ANT2. Oncogene 2016; 35(10):1236-49; http://dx.doi.org/10.1038/onc.2015.162
- Kim YK, Bae GU, Kang JK, Park JW, Lee EK, Lee HY, Choi WS, Lee HW, Han JW. Cooperation of H2O2-mediated ERK activation with Smad pathway in TGF-beta1 induction of p21WAF1/Cip1. Cellular Signal 2006; 18:236-43; PMID:15979845; http://dx.doi.org/10.1016/j.cellsig.2005.04.008
- Hubackova S, Krejcikova K, Bartek J, Hodny Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging 2012; 4:932-51; PMID:23385065; http://dx.doi.org/10.18632/aging.100520
- Marlow R, Honeth G, Lombardi S, Cariati M, Hessey S, Pipili A, Mariotti V, Buchupalli B, Foster K, Bonnet D, et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res 2013; 73:6886-99; PMID:24145351; http://dx.doi.org/10.1158/0008-5472.CAN-13-0991
- Braun S, Pantel K, Muller P, Janni W, Hepp F, Kentenich CR, Gastroph S, Wischnik A, Dimpfl T, Kindermann G, et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N Engl J Med 2000; 342:525-33; PMID:10684910; http://dx.doi.org/10.1056/NEJM200002243420801
- Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, MacDonald IC. Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 2000; 60:2541-6; PMID:10811137
- Goodison S, Kawai K, Hihara J, Jiang P, Yang M, Urquidi V, Hoffman RM, Tarin D. Prolonged dormancy and site-specific growth potential of cancer cells spontaneously disseminated from nonmetastatic breast tumors as revealed by labeling with green fluorescent protein. Clin Cancer Res 2003; 9:3808-14; PMID:14506175
- Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, Morris VL, Groom AC, Chambers AF. Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Res 2002; 62:2162-8; PMID:11929839
- Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, Shen R, Brogi E, Brivanlou AH, Giancotti FG. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012; 150:764-79; PMID:22901808; http://dx.doi.org/10.1016/j.cell.2012.06.035
- Ruppender N, Larson S, Lakely B, Kollath L, Brown L, Coleman I, Coleman R, Nguyen H, Nelson PS, Corey E, et al. Cellular adhesion promotes prostate cancer cells escape from dormancy. PloS One 2015; 10:e0130565; PMID:26090669; http://dx.doi.org/10.1371/journal.pone.0130565
- Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A 2009; 106:10290-5; PMID:19502425; http://dx.doi.org/10.1073/pnas.0904227106
- Morgan TM, Lange PH, Porter MP, Lin DW, Ellis WJ, Gallaher IS, Vessella RL. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clin Cancer Res 2009; 15:677-83; PMID:19147774; http://dx.doi.org/10.1158/1078-0432.CCR-08-1754
- He W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MA, Massague J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell 2006; 125:929-41; PMID:16751102; http://dx.doi.org/10.1016/j.cell.2006.03.045