
ABSTRACT
The increased propensity of BRCA1 mutation carriers to develop aggressive breast tumors with stem-like properties begins to be understood in terms of osteoprotegerin (OPG)-unrestricted cross-talk between RANKL-overproducing progesterone-sensor cells and cancer-initiating RANK+ responder cells that reside within pre-malignant BRCA1mut/+ breast epithelial tissue. We recently proposed that, in the absence of hormone influence, cancer-initiating cells might remain responsive to RANKL stimulation, and hence to the therapeutic effects of the anti-RANKL antibody denosumab because genomic instability induced by BRCA1 haploinsufficiency might suffice to cell-autonomously hyperactivate RANKL gene expression. Here we report that the biguanide metformin prevents BRCA1 haploinsufficiency-driven RANKL gene overexpression, thereby disrupting an auto-regulatory feedback control of RANKL-addicted cancer stem cell-like states within BRCA1mut/− cell populations. Moreover, metformin treatment elicits a synergistic decline in the breast cancer-initiating cell population and its self-renewal capacity in BRCA1-mutated basal-like breast cancer cells with bone metastasis-initiation capacity that exhibit primary resistance to denosumab in mammosphere assays. The specific targeting of RANKL/RANK signaling with denosumab is expected to revolutionize prevention and treatment strategies currently available for BRCA1 mutation carriers. Our findings provide a rationale for new denosumab/metformin combinatorial strategies to clinically manage RANKL-related breast oncogenesis and metastatic progression.
The cytokines receptor activator of nuclear factor κB ligand (RANKL) and osteoprotegerin (OPG) play key roles in the differentiation and function of osteoblasts, cells that are specialized in the degradation of bone tissue.Citation1-3 Binding of RANKL to its receptor, RANK, on the cell surface of osteoclasts or their precursors induces differentiation, activation, and survival, and stimulates bone resorption. OPG is the endogenous decoy receptor for RANKL that antagonizes RANKL/RANK-mediated signaling and hence inhibits bone resorption, preserving bone density. The balance between RANKL and OPG is important for bone metabolism and its dysregulation is a major driving force in cancer treatment-induced bone loss (CTIBL) and in the development of malignant bone lesions.Citation4 Not surprisingly, the RANKL/OPG signaling axis has emerged as an attractive target not only to prevent bone loss, but also to prevent bone metastases in breast cancer.Citation5,6
Denosumab, a fully human antibody targeting RANKL, is a paradigmatic example of the family of bone resorption inhibitors developed to treat osteoporosis and to prevent skeletal damage caused by bone metastases. Bisphosphonates such as zoledronic acid are the current standard of care for the treatment of skeletal-related events (SREs), including bone metastases, in patients with advanced breast cancer.Citation3,4 However, results from clinical studies suggest that denosumab efficacy, as determined by a reduction in SREs, is at least equal to bisphosphonates and possibly superior in a large prospectively randomized phase III trial of advanced breast cancer patients.Citation3-5 Ongoing clinical studies evaluating the effects of denosumab in CTIBL and on bone metastasis-free survival in women with early-stage breast cancer at high risk for disease recurrence should confirm whether it can challenge the bisphosphonate-based current standard care of bone metastasis in breast cancer patients. The repurposing of an osteoclastogenesis inhibitor such as denosumab may herald a new era of primary prevention therapies, and perhaps cancer treatments, in woman carriers of BRCA1 gene mutations.
Dysregulation of the OPG/RANKL/RANK signaling axis appears to play a role in breast carcinogenesis that may be particularly relevant for BRCA1-mutation carriers facing a high lifetime risk of breast cancer.Citation7-10 The increased propensity of this population to develop aggressive breast tumors with stem-like properties begins to be understood in terms of OPG-unrestricted cross-talk between RANKL-overproducing cells and cancer-initiating RANK+ cells that reside within pre-malignant breast epithelial tissues.Citation11,12 Free serum levels of OPG are significantly lower in BRCA1 mutation carriers than in non-carrier controls. Moreover, among BRCA1 carriers, lower OPG levels are associated with germline mutations known to confer an increased risk for breast cancer. High plasma OPG levels in BRCA1-mutation carriers correlate with a significantly decreased risk of developing breast cancer as compared with BRCA1-deficient women with low OPG levels.Citation7-9 These findings point to a promising role for OPG as a biomarker to identify BRCA1 mutation carriers who are at higher risk of developing breast cancer and who would benefit from therapies mimicking the physiological role of OPG, such as the anti-RANKL antibody denosumab.Citation10
The net magnitude of the OPG/RANKL/RANK signaling axis in driving BRCA1-related breast carcinogenesis seems to be determined by the decrease in local and systemic OPG along with the local increase of RANKL, thus supporting the relevance of therapeutically manipulating OPG levels in BRCA1 carriers by directly targeting RANKL. Along this line, 2 highly publicized studies have recently reported that RANKL inhibition, achieved by using either a recombinant OPG-Fc fusion protein or RANKL-specific monoclonal antibodies such as denosumab, can significantly delay tumor onset in clinically relevant models of BRCA1 deficiency.Citation11, 12 Mechanistically, denosumab is thought to interfere with the cross-talk between RANKL-producing “sensor” cells and cancer-initiating RANK+ responder cells that appear to reside within pre-malignant tissues of BRCA1-mutation carriers. We recently provided support to the alternative but not mutually exclusive hypothesis that BRCA1 haploinsufficiency may be sufficient to cell-autonomously activate RANKL expression and generate breast cancer-initiating cell populations responsive to denosumab.Citation13
Metformin prevents BRCA1 haploinsufficiency-driven upregulation of RANKL in breast epithelial cells. The anti-diabetic biguanide metformin is consistently associated with a decrease in cancer incidence and mortality, both overall and also linked to specific organ sites including breast.Citation14-16 Metformin has also been shown to exert direct osteogenic effects by enhancing osteoblast differentiation and inhibiting osteoclast differentiation in vitro and by preventing bone loss in ovariectomized rats.Citation17-21 The anti-osteoclastogenic activity of metformin seems to involve the stimulation of OPG production and the reduction of RANKL expression by osteoblasts. We recently provided unbiased evidence that mono-allelic inactivation of BRCA1 in normal-like breast epithelial cells suffices to cell-autonomously generate RANKL-overproducing, denosumab-responsive cancer stem cell (CSC)-like cellular states.Citation13 Thus, we envisioned that the unanticipated capacity of metformin to regulate breast epithelia-associated RANKL expression might be a new approach to target RANKL/RANK signaling that generates and maintains CSC-like properties in BRCA1-deficient breast cancer cells.
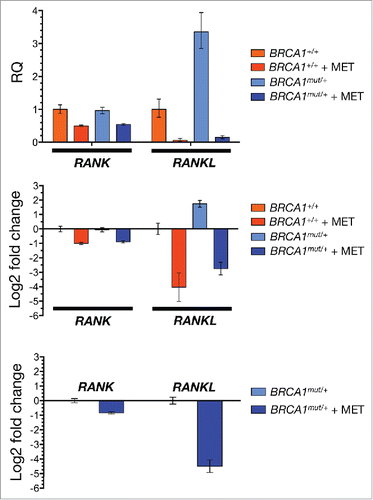
We recently reported activation of RANKL gene expression in normal-like MCF10A human breast epithelial cells containing the 185delAG mutation in one BRCA1 allele.Citation13 This cell line is a good model of genomic instability and recapitulates the cell-autonomous consequences of one-hit BRCA1 inactivation in breast epithelia of carriers of BRCA1mutations.Citation22-24 We here assessed the impact of metformin treatment on the expression status of RANK and RANKL in BRCA1+/+ parental cells and in BRCA1185delAG/+ haploinsufficient isogenic derivatives. RT-PCR analysis showed that metformin treatment (5 mmol/L) moderately reduced the expression of RANK in both cell lines (). Remarkably, the ability of metformin to substantially reduce RANKL gene expression in BRCA1+/+ parental cells (4-fold reduction vs. untreated BRCA1+/+ cells) remained unaltered in a background of BRCA1 deficiency. Accordingly, the very high levels of RANKL gene expression triggered by BRCA1 haploinsufficiency were completely suppressed (5-fold reduction vs. untreated BRCA1185delAG/+ cells) by metformin (). These findings reveal for the first time that metformin might target the hyperactivation of the RANKL/RANK signaling axis by preventing BRCA1 deficiency-induced cell-autonomous hyperactivation of RANKL gene expression.
Figure 1. Metformin inhibits RANKL gene expression in breast epithelial cells. Total RNA from untreated and metformin-treated (5 mmol/L; 48 h) BRCA1+/+ and BRCA1mut/+ MCF10A isogenic cell pairs was evaluated in technical triplicates for the abundance of RANK (TNFRSF11A, Hs00921372_m1) and RANKL (TNFSF11, Hs00243522_m1) relative to housekeeping genes GADPH (Hs99999905_m1) and 18S (Hs99999901_s1). The transcript abundance was calculated using the delta Ct method and presented as relative quantification (RQ) or log2 fold-change, as specified. MET, Metformin.
Metformin synergistically sensitizes BRCA1-deficient breast cancer stem cells to denosumab. Our previous findings showing the robust ability of metformin to reduce the mammosphere-initiating capacity of BRCA1185delAG/+ cells did not include an analysis of the nature of the interaction between metformin and denosumab.Citation13, 24 Thus, we sought to explore the therapeutic relevance of metformin for the RANKL-dependent ability of denosumab to reduce tumor-initiating capacity of triple-negative MDA-MB-436 cells. Metastatic MDA-MB-436 cells contain a BRCA1 5396 + 1G>A mutation, resulting in an in-frame deletion of 28 amino acids and a truncated BRCA1 protein,Citation25 and they are capable of developing osteolytic mammary tumors upon direct intra-tibial injection into immunocompromised mice.Citation26, 27 Since the number and the size of mammospheres reflects the quantity of CSC-like cells capable of self-renewal and the self-renewal capacity of each CSC-like cell, respectively, we used the recently developed Cell2Sphere™ Kit (StemTek Therapeutics, Bilbao, Spain) to evaluate the impact of denosumab and metformin on the number and size of mammospheres formed by BRCA1-mutant MDA-MB-436 cells. Treatment with either a clinically relevant concentration of denosumab [1 μg/mL; 28] or metformin (1 mmol/L) as single agents moderately reduced (∼20% and 10%, respectively) mammosphere-forming efficiency (MSFE) of MDA-MB-436 cells (). Remarkably, the concurrent combination of denosumab and metformin supra-additively decreased MSFE by greater than 50%. Moreover, under these conditions mammospheres were markedly smaller than those generated from untreated control cells; indeed, there were no mammospheres >100 μm in size ().
Figure 2. Metformin sensitizes mammosphere-initiating cells to denosumab. Cell2Sphere™ assays using BRCA1-deficient MDA-MB-436 cells were performed as per the manufacturer's instructions ((http://stemtektherapeutics.com/en/cell2sphere#cell2sphere_kit). Drugs were added to quintuplicate sets of wells on days 1 and 4 without replenishing the medium. ImageJ was used to quantify the size (left; central lines indicate mean values) and number (right) of 6-day-old mammospheres. *P < 0.05, **P < 0.001 MET, Metformin. Size bar = 2000 μm.
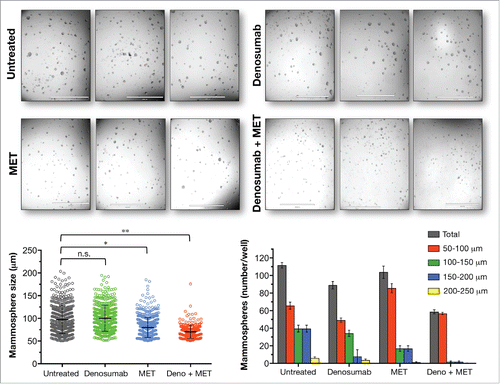
The statistically significant reduction in the number and size of mammospheres generated with denosumab plus metformin but not with denosumab alone indicates that metformin functions synergistically to decrease the magnitude of the mammosphere-initiating population as well as its self-renewal capacity in denosumab-refractory CSC-like cells.
Metformin-regulated RANKL: From cancer prevention to circumventing denosumab resistance. Beyond prophylactic mastectomy, there are currently few options available to reduce the risk of developing breast cancer for BRCA1 mutation carriers.Citation23 We are rapidly accumulating evidence that susceptibility to oncogenesis and tissue specificity in BRCA1 mutation carriers is, at least in part, a net consequence of cell nonautonomous and cell-autonomous factors of the OPG/RANKL/RANK axis.
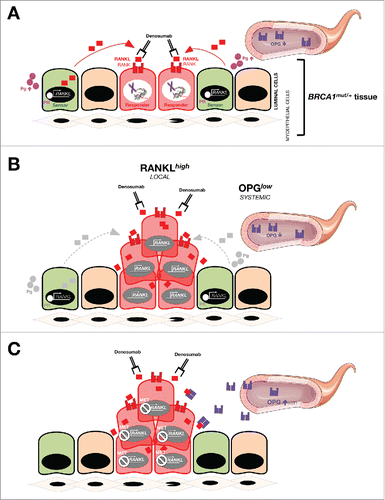
Whereas aberrant DNA damage-triggered hyperactivation of the RANKL/NF-κB signaling axis might account for the expansion of cell populations within BRCA1mut/+ pre-neoplastic breast tissue with breast cancer-initiating capability, low systemic levels of the natural antagonist OPG would provoke an unrestrained amplification of such genetically unstable populations of BRCA1 haploinsufficient cells with ensuing DNA damage activation of RANKL/NF-κB signaling. Such a model of epithelium hyperproliferation during breast oncogenesis in pre-menopausal BRCA1 mutation carriers likely involves a progesterone-inducible secretion of RANKL in so-called sensor cells (i.e., mature ductal cells) and subsequent RANKL stimulation of so-called “responder” breast cancer-initiating RANK+ cells (i.e., hormone receptor-negative luminal progenitors).Citation29 Our recent finding that triple-negative/basal-like breast epithelial cells, engineered to have impaired DNA repair and consequent genomic instability upon targeted mutational knockout of BRCA1 single allele, overproduce RANKL strongly suggests that RANKL-addicted cell populations with breast cancer-initiating capacity would persist during hyperplasia, even in the absence of hormonal influence and despite the drop in RANKL expression occurring in the post-menopausal setting.Citation12 An idoneous approach for breast cancer prevention might therefore involve strategies to revert the chronically aberrant high RANKL:OPG ratio in breast epithelium of BRCA1-mutation carriers by increasing systemic (serum) OPG while decreasing local RANKL in epithelial cells ().
Figure 3. Dysregulation of the OPG/RANKL/RANK signaling axis and BRCA1 deficiency-driven breast oncogenesis: (A)new target for anti-cancer metformin. Molecular dialog between progesterone (Pg)-responsive cells and hormone receptor (HR)-negative luminal progenitors seems to drive the hyperproliferation of breast cancer-initiating cell populations within the breast epithelia of BRCA1 mutation carriers.Citation11,12 In a hormone-dependent stage (e.g., pre-menopausal women), RANKL secretion might become stimulated upon binding of Pg to its receptor (PR) in mature ductal cells (i.e., so-called “sensor” cells); secreted RANKL then binds and activates RANK on RANK+ luminal progenitors (i.e., so-called “responder” cells). Generally, OPG negatively regulates RANKL/RANK signaling by binding to RANKL as an endogenous decoy receptor, thereby inhibiting signal transduction. However, the mitogenic cross-talk between RANKL-producing sensor cells and RANKL-addicted responder cells might be less restricted in high-risk subgroups of BRCA1 mutation carriers with significant lower plasma levels of OPG. The net magnitude of the OPG/RANKL/RANK signaling axis in driving BRCA1-related breast oncogenesis may be largely determined by local increases of RANKL along with decreases in systemic OPG. Our recent findings in HR-negative, basal-like MCF10A breast epithelial cells with a pathogenic185delAG mutation in one BRCA1 alleleCitation13 further suggest that, in the absence of hormone influence, breast cancer-initiating cells may remain responsive to RANKL stimulation because compromised DNA repair and consequent genomic instability provoked by BRCA1 haploinsufficiency might suffice to establish hyperplasic states via an auto-regulatory feedback loop triggered by cell-autonomous hyperactivation of RANKL gene expression. Such a hormone-independent scenario might become reinforced by low circulating levels of OPG not only in post-menopausal BRCA1 carriers but also in other post-menopausal groups at high risk for breast cancer and in women with circulating tumor cells. The ability of metformin (MET) to restore dysregulated OPG/RANKL/RANK signaling may represent an important clinical opportunity to prevent breast cancer and maintain bone health (e.g., following salpingo-oophorectomy) in BRCA mutation carriers and to treat RANKL-driven metastasis-initiating lesions in combination with denosumab.
Preclinical models of inherited cancer syndromes are increasingly appreciated as valuable experimental systems to develop chemopreventive agents (e.g., denosumab and metformin) for application in hereditary and sporadic cancer settings.Citation30 While it is acknowledged that the potential of denosumab as a non-surgical preventive therapy to delay or prevent BRCA1-related breast cancer should await the results of large randomized clinical trials, the ability of metformin to potentially lower the RANKL/OPG ratio, by inhibiting local RANKL gene expression and/or increasing systemic levels of OPG, warrants further exploration as a combinatorial strategy that might prove safe and effective for pre-menopausal BRCA1 mutation carriers before they consider mastectomy. Such a combination could potentially have broader applicability, including post-menopausal BRCA1 carriers and others at increased genetic or familial risk. Moreover, because metformin has been shown to significantly increase total body bone mineral density and prevent bone loss in ovariectomized adult rats,Citation19 its ability to affect bone metabolismCitation31 together with the RANKL-targeting activities of metformin and denosumab might synergize to minimize the loss in bone mineral density that accompanies ovarian cancer-preventive bilateral salpingo-oophorectomy in BRCA mutation carriers.
The ability of denosumab to reduce mammosphere forming capacity--an accepted surrogate for the relative proportion and survival of CSCs--not only in BRCA1 haploinsufficient breast epithelial cells but also in triple-negative/basal-like and HER2-overexpressing breast cancer cells,Citation13 supports the notion that tumor- and metastasis-initiating cells from genetically diverse breast cancer subtypes might share a common addiction to hyperactive RANKL signaling in primary and metastatic locations including the bone microenvironment.Citation32 Beyond prevention, the exploration of a role for therapeutic targeting of RANKL with denosumab for the treatment of established breast cancer might take advantage of the expected incorporation of new biomarkers capable of stratifying breast cancer patients that will benefit from bone-targeting, metastasis-preventing treatments such as denosumab from those that, because of de novo or acquired mechanisms of escape, require alternative or additional sensitizing therapies.Citation33 Triple-negative/basal-like MDA-MB-436 cells with well-recognized bone metastasis-initiation capacityCitation26, 27 were found to behave as denosumab-resistant breast cancer cells in terms of tumorsphere-initiating capacity. Remarkably, they became sensitized to the RANKL-targeting activity of denosumab in the presence of metformin. Intriguingly, denosumab-resistant MDA-MB-436 cells exhibit a high copy number of the proto-oncogene c-MAF,Citation34 a recently discovered bone metastasis biomarker that might help to distinguish responders and non-responders to RANKL-targeted therapies.Citation35
The specific targeting of autocrine/paracrine RANKL/RANK signaling with denosumab is expected to revolutionize prevention and treatment strategies currently available for BRCA1 mutation carriers. We propose that the ability of metformin to: a) suppress breast epithelial RANKL gene expression; b) impair the breast cancer-initiating capacity of multiple breast cancer subtypes including those of cancer-prone BRCA1mut/+ haploinsufficient stages,Citation24, 36-38 and c.) synergize with the RANKL-dependent actions of denosumab in breast cancer cells with bone metastasis-initiation abilities, may provide a strong rationale for new denosumab/metformin combinatorial strategies to clinically manage RANKL-related breast oncogenesis and metastatic progression.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
The authors would like to thank Dr. Kenneth McCreath for editorial support.
Funding
This work was supported by grants from the Ministerio de Ciencia e Innovación (Grant SAF2016–80639-P to J. A. Menendez), Plan Nacional de I+D+I, Spain and the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) (Grant 2014 SGR229 to J. A. Menendez), Departament d'Economia i Coneixement, Catalonia, Spain. Elisabet Cuyàs is supported by the Sara Borrell post-doctoral contract (CD15/00033) from the Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), Spain. Joaquim Bosch-Barrera is supported by an Emerging Research Grant (2013) from the Spanish Society of Medical Oncology (SEOM, Madrid, Spain), and a Research Grant from Pfizer (WI190764).
References
- Kobayashi Y, Udagawa N, Takahashi N. Action of RANKL and OPG for osteoclastogenesis. Crit Rev Eukaryot Gene Expr 2009; 19:61-72; PMID:19191757; https://doi.org/10.1615/CritRevEukarGeneExpr.v19.i1.30
- Martin TJ, Sims NA. RANKL/OPG; Critical role in bone physiology. Rev Endocr Metab Disord 2015; 16:131-9; PMID:25557611; https://doi.org/10.1007/s11154-014-9308-6
- Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, San Martin J, Dansey R. Bench to bedside: elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat Rev Drug Discov 2012; 11:401-19; PMID:22543469; https://doi.org/10.1038/nrd3705
- Brown-Glaberman U, Stopeck AT. Impact of denosumab on bone mass in cancer patients. Clin Pharmacol 2013; 5:117-29; PMID:23861604.
- Gül G, Sendur MA, Aksoy S, Sever AR, Altundag K. A comprehensive review of denosumab for bone metastasis in patients with solid tumors. Curr Med Res Opin 2016; 32:133-45; PMID:26451465; https://doi.org/10.1185/03007995.2015.1105795
- Dougall WC, Holen I, González Suárez E. Targeting RANKL in metastasis. Bonekey Rep 2014; 3:519; PMID:24795813; https://doi.org/10.1038/bonekey.2014.14
- Widschwendter M, Burnell M, Fraser L, Rosenthal AN, Philpott S, Reisel D, Dubeau L, Cline M, Pan Y, Yi PC, Gareth Evans D, Jacobs IJ, Menon U, Wood CE, Dougall WC. Osteoprotegerin (OPG), The endogenous inhibitor of receptor activator of NF-κB Ligand (RANKL), is Dysregulated in BRCA mutation carriers. EBioMedicine 2015; 2:1331-9; PMID:26629528; https://doi.org/10.1016/j.ebiom.2015.08.037
- Odén L, Akbari M, Zaman T, Singer CF, Sun P, Narod SA, Salmena L, Kotsopoulos J. Plasma osteoprotegerin and breast cancer risk in BRCA1 and BRCA2 mutation carriers. Oncotarget 2016; 7:86687-86694; PMID:27893411.
- Kiechl S, Schramek D, Widschwendter M, Fourkala EO, Zaikin A, Jones A, Jaeger B, Rack B, Janni W, Scholz C, Willeit J, Weger S, Mayr A, Teschendorff A, Rosenthal A, Fraser L, Philpott S, Dubeau L, Keshtgar M, Roylance R, Jacobs IJ, Menon U, Schett G, Penninger JM. Aberrant regulation of RANKL/OPG in women at high risk of developing breast cancer. Oncotarget 2017; 8:3811-3825; PMID:28002811.
- Kotsopoulos J, Singer C, Narod SA. Can we prevent BRCA1-associated breast cancer by RANKL inhibition?. Breast Cancer Res Treat 2017; 161:11-16; PMID:27783278; https://doi.org/10.1007/s10549-016-4029-z
- Sigl V, Owusu-Boaitey K, Joshi PA, Kavirayani A, Wirnsberger G, Novatchkova M, Kozieradzki I, Schramek D, Edokobi N, Hersl J, Sampson A, Odai-Afotey A, Lazaro C, Gonzalez-Suarez E, Pujana MA, Cimba F, Heyn H, Vidal E, Cruickshank J, Berman H, Sarao R, Ticevic M, Uribesalgo I, Tortola L, Rao S, Tan Y, Pfeiler G, Lee EY, Bago-Horvath Z, Kenner L, Popper H, Singer C, Khokha R, Jones LP, Penninger JM. RANKL/RANK control Brca1 mutation-driven mammary tumors. Cell Res 2016; 26:761-74; PMID:27241552; https://doi.org/10.1038/cr.2016.69
- Nolan E, Vaillant F, Branstetter D, Pal B, Giner G, Whitehead L, Lok SW, Mann GB, Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab), Rohrbach K, Huang LY, Soriano R, Smyth GK, Dougall WC, Visvader JE, Lindeman GJ. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med 2016; 22:933-9; PMID:27322743; https://doi.org/10.1038/nm.4118
- Cuyàs E, Corominas-Faja B, Muñoz-San Martín M, Martin-Castillo B, Lupu R, Brunet J, Bosch-Barrera J, Menendez JA. BRCA1 haploinsufficiency cell-autonomously activates RANKL expression and generates denosumab-responsive breast cancer-initiating cells. Oncotarget 2017; [epub ahead of print]; PMID:28388533; https://doi.org/10.18632/oncotarget.16558
- Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B, Menendez JA. Metformin: multi-faceted protection against cancer. Oncotarget 2011; 2:896-917; PMID:22203527; https://doi.org/10.18632/oncotarget.387
- Chae YK, Arya A, Malecek MK, Shin DS, Carneiro B, Chandra S, Kaplan J, Kalyan A, Altman JK, Platanias L, Giles F. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget 2016; 7:40767-40780; PMID:27004404.
- Menendez JA, Martin-Castillo B, Joven J. Metformin and cancer: Quo vadis et cui bono?. Oncotarget 2016; 7:54096-54101.
- Cortizo AM, Sedlinsky C, McCarthy AD, Blanco A, Schurman L. Osteogenic actions of the anti-diabetic drug metformin on osteoblasts in culture. Eur J Pharmacol 2006; 536:38-46; PMID:16564524; https://doi.org/10.1016/j.ejphar.2006.02.030
- Lee YS, Kim YS, Lee SY, Kim GH, Kim BJ, Lee SH, Lee KU, Kim GS, Kim SW, Koh JM. AMP kinase acts as a negative regulator of RANKL in the differentiation of osteoclasts. Bone 2010; 47:926-37; PMID:20696287; https://doi.org/10.1016/j.bone.2010.08.001
- Mai QG, Zhang ZM, Xu S, Lu M, Zhou RP, Zhao L, Jia CH, Wen ZH, Jin DD, Bai XC. Metformin stimulates osteoprotegerin and reduces RANKL expression in osteoblasts and ovariectomized rats. J Cell Biochem 2011; 112:2902-9; PMID:21618594; https://doi.org/10.1002/jcb.23206
- Liu L, Zhang C, Hu Y, Peng B. Protective effect of metformin on periapical lesions in rats by decreasing the ratio of receptor activator of nuclear factor kappa B ligand/osteoprotegerin. J Endod 2012; 38:943-7; PMID:22703658; https://doi.org/10.1016/j.joen.2012.03.010
- Wang J, Chen TY, Qin S, Duan Y, Wang G. Inhibitory effect of metformin on bone metastasis of cancer via OPG/RANKL/RANK system. Med Hypotheses 2013; 81:805-6; PMID:24074896; https://doi.org/10.1016/j.mehy.2013.08.032
- Konishi H, Mohseni M, Tamaki A, Garay JP, Croessmann S, Karnan S, Ota A, Wong HY, Konishi Y, Karakas B, Tahir K, Abukhdeir AM, Gustin JP, Cidado J, Wang GM, Cosgrove D, Cochran R, Jelovac D, Higgins MJ, Arena S, Hawkins L, Lauring J, Gross AL, Heaphy CM, Hosokawa Y, Gabrielson E, Meeker AK, Visvanathan K, Argani P, Bachman KE, Park BH. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc Natl Acad Sci U S A 2011; 108:17773-8; PMID:21987798; https://doi.org/10.1073/pnas.1110969108
- Menendez JA, Folguera-Blasco N, Cuyàs E, Fernández-Arroyo S, Joven J, Alarcón T. Accelerated geroncogenesis in hereditary breast-ovarian cancer syndrome. Oncotarget 2016; 7:11959-71; PMID:26943589.
- Cuyàs E, Fernández-Arroyo S, Alarcón T, Lupu R, Joven J, Menendez JA. Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: evidence for metformin-based “starvation” strategies in BRCA1 carriers. Oncotarget 2016; 7:52974-52992; PMID:27259235.
- Elstrodt F, Hollestelle A, Nagel JH, Gorin M, Wasielewski M, van den Ouweland A, Merajver SD, Ethier SP, Schutte M. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res 2006; 66:41-5; PMID:16397213; https://doi.org/10.1158/0008-5472.CAN-05-2853
- Ottewell PD, Woodward JK, Lefley DV, Evans CA, Coleman RE, Holen I. Anticancer mechanisms of doxorubicin and zoledronic acid in breast cancer tumor growth in bone. Mol Cancer Ther 2009; 8:2821-32; PMID:19789217; https://doi.org/10.1158/1535-7163.MCT-09-0462
- Wright LE, Ottewell PD, Rucci N, Peyruchaud O, Pagnotti GM, Chiechi A, Buijs JT, Sterling JA. Murine models of breast cancer bone metastasis. Bonekey Rep 2016; 5:804; PMID:27867497; https://doi.org/10.1038/bonekey.2016.31
- Sohn W, Simiens MA, Jaeger K, Hutton S, Jang G. The pharmacokinetics and pharmacodynamics of denosumab in patients with advanced solid tumours and bone metastases: A systematic review. Br J Clin Pharmacol 2014; 78:477-87; PMID:24548274; https://doi.org/10.1111/bcp.12355
- Nolan E, Lindeman GJ, Visvader JE. Out-RANKing BRCA1 in mutation carriers. Cancer Res 2017; 77:595-600; PMID:28104682; https://doi.org/10.1158/0008-5472.CAN-16-2025
- Walcott FL, Patel J, Lubet R, Rodriguez L, Calzone KA. Hereditary cancer syndromes as model systems for chemopreventive agent development. Semin Oncol 2016; 43:134-45; PMID:26970132; https://doi.org/10.1053/j.seminoncol.2015.09.015
- McCarthy AD, Cortizo AM, Sedlinsky C. Metformin revisited: Does this regulator of AMP-activated protein kinase secondarily affect bone metabolism and prevent diabetic osteopathy. World J Diabetes 2016; 7:122-33; PMID:27022443; https://doi.org/10.4239/wjd.v7.i6.122
- Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, Quraishi AA, Ignatoski KW, Daignault S, Davis A, Hall CL, Palanisamy N, Heath AN, Tawakkol N, Luther TK, Clouthier SG, Chadwick WA, Day ML, Kleer CG, Thomas DG, Hayes DF, Korkaya H, Wicha MS. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res 2013; 73:1635-46; PMID:23442322; https://doi.org/10.1158/0008-5472.CAN-12-3349
- Coleman RE. Bone cancer in 2011: Prevention and treatment of bone metastases. Nat Rev Clin Oncol 2011; 9:76-8; PMID:22182971; https://doi.org/10.1038/nrclinonc.2011.198
- Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J, Pau G, Reeder J, Cao Y, Mukhyala K, Selvaraj SK, Yu M, Zynda GJ, Brauer MJ, Wu TD, Gentleman RC, Manning G, Yauch RL, Bourgon R, Stokoe D, Modrusan Z, Neve RM, de Sauvage FJ, Settleman J, Seshagiri S, Zhang Z. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 2015; 33:306-12; PMID:25485619; https://doi.org/10.1038/nbt.3080
- Pavlovic M, Arnal-Estapé A, Rojo F, Bellmunt A, Tarragona M, Guiu M, Planet E, Garcia-Albéniz X, Morales M, Urosevic J, Gawrzak S, Rovira A, Prat A, Nonell L, Lluch A, Jean-Mairet J, Coleman R, Albanell J, Gomis RR. Enhanced MAF oncogene expression and breast cancer bone metastasis. J Natl Cancer Inst 2015; 107(12):djv256; PMID:26376684; https://doi.org/10.1093/jnci/djv256
- Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat 2011; 126:355-64; PMID:20458531; https://doi.org/10.1007/s10549-010-0924-x
- Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 2010; 9:3807-14; PMID:20890129; https://doi.org/10.4161/cc.9.18.13131
- Cufi S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, Martin-Castillo B, Menendez JA. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget 2012; 3:395-8; PMID:22565037; https://doi.org/10.18632/oncotarget.488