
ABSTRACT
Telomeres are nucleoprotein structures that cap the ends of linear chromosomes. Telomere homeostasis is central to maintaining genomic integrity. In budding yeast, Cdk1 phosphorylates the telomere-specific binding protein, Cdc13, promoting the recruitment of telomerase to telomere and thereby telomere elongation. Cdc13 is also an integral part of the CST (Cdc13-Stn1-Ten1) complex that is essential for telomere capping and counteracting telomerase-dependent telomere elongation. Therefore, telomere length homeostasis is a balance between telomerase-extendable and CST-unextendable states. In our earlier work, we showed that Cdk1 also phosphorylates Stn1 which occurs sequentially following Cdc13 phosphorylation during cell cycle progression. This stabilizes the CST complex at the telomere and results in telomerase inhibition. Hence Cdk1-dependent phosphorylations of Stn1 acts like a molecular switch that drives Cdc13 to complex with Stn1-Ten1 rather than with telomerase. However, the underlying mechanism of how a single cyclin-dependent kinase phosphorylates Cdc13 and Stn1 in temporally distinct windows is largely unclear. Here, we show that S phase cyclins are necessary for telomere maintenance. The S phase and mitotic cyclins facilitate Cdc13 and Stn1 phosphorylation respectively, to exert opposing outcomes at the telomere. Thus, our results highlight a previously unappreciated role for cyclins in telomere replication.
Introduction
Telomeres are nucleoprotein structures found at the ends of eukaryotic linear chromosomes that protect chromosome ends from being detected as DNA double stranded breaks (DSBs), thereby maintaining genomic integrity.Citation1-4 Telomeres consist of double-stranded TG repeat sequences in tandem followed by a short single-stranded (ss) 3′ G-rich overhang.Citation5-7 Specific proteins bind to the single and double stranded regions of the telomeres, together forming a protective nucleoprotein cap at the chromosomal ends (reviewed in Citation8). Telomere replication is coupled to DNA replication in which the leading and lagging strand synthesis of the telomeres are closely coordinated. After replication by conventional DNA polymerases, the removal of terminal RNA primer from the lagging strand results in an 8–12 base gap at the 5′ end of the newly synthesized DNA molecules. On the other hand, leading strand synthesis terminates in blunt end, which undergoes end processing to result in terminal single-stranded overhang. In the absence of a mechanism to maintain telomere length, this shortening of telomeres as a result of semi-conservative DNA replication, known as the end replication problem, leads to progressive terminal attrition of the chromosomal ends through successive cell division cycles and eventually results in critically short telomeres that trigger cell cycle arrest and senescence.Citation9-11 To counteract the end replication problem, the enzyme telomerase, a unique ribonucleoprotein, adds telomeric repeats to chromosomal ends and ensures maintenance of telomeres.Citation12 The telomerase holoenzyme consists of a catalytic subunit and an integral RNA template which it uses to lengthen the G-rich strand of the telomeric DNA.Citation13 The telomerase-dependent telomere elongation is closely coordinated with C-strand fill-in and subject to exonucleolytic post-processing to result in newly synthesized telomeres that are similar in length to its parental strands.Citation14 The balance between telomere elongation and attrition is fundamental to telomere length homeostasis. Disruption of telomere homeostasis leads to several diseases including cancers and premature aging syndromes in humans (reviewed in Citation15,16). Over 85–90% of human cancers have upregulated levels of telomerase that confer unlimited proliferative potential to the cancer cells.Citation17-19 On the other hand, telomere syndromes such as coats plus, dyskeratosis congenita, aplastic anemia and idiopathic pulmonary fibrosis, occur due to deregulated telomere maintenance, causing premature aging especially in the stem cells of telomere syndrome patients.Citation20-25
In budding yeast, telomere replication is largely regulated by Cyclin-dependent kinase 1 (Cdk1), also known as Cdc28, and is restricted to the late S to G2 phase of the cell cycle.Citation26 Cdk1 is central to orchestrating telomere replication, elongation and maintenance by regulating the temporal recruitment of telomerase and the Cdc13-Stn1-Ten1 (CST) complex to telomeres during cell cycle progression.Citation27,28 Furthermore, Cdk1 also controls telomere end processing and C-strand fill-in through its regulation on Exo1 nuclease and DNA Polymerase α, respectively.Citation29,30 The telomerase holoenzyme in yeast consists of the catalytic subunit Est2, the RNA template TLC1 and subunits Est1 and Est3.Citation31-33 The budding yeast telomere, about 250–350 bp in length, is bound by Rap1-Rif1-Rif2 complexes on its double-stranded region and by the CST complexes on its single-stranded tail. The CST complex is a telomere-specific Replication Protein A-like complex that plays a central role in telomere homeostasis and chromosomal end protection through its telomere capping and telomerase inhibition functions.Citation34-37 Cdc13, an integral component of the CST complex, also interacts with Est1 of the telomerase holoenzyme and this interaction promotes the recruitment of telomerase to the telomeres while its disruption causes progressive shortening of telomeres that eventually compromises cell viability.Citation34,38 Loss of Cdc13 not only results in short telomeres but also causes telomere uncapping, accumulation of single-stranded G-rich overhang and DNA damage checkpoint activation that are consistent with Cdc13s role in telomere protection.Citation39,40 Cdc13 exerts its telomere capping functions through its interaction with Stn1 and Ten1, forming a stable CST complex that is central to protecting chromosomal ends from the DNA damage repair machinery as loss of these telomeric proteins triggers DNA damage signaling and checkpoint activation leading to cell cycle arrest at G2/M.Citation28,35,36,39,41-46 The CST complex also inhibits uncontrolled Exo1-dependent 5′ to 3′ resection of telomere to prevent excessive accumulation of single-stranded G-overhangs that triggers DNA damage response. Thus, the CST complex closely coordinates with DNA damage response machinery for telomere end processing Citation47-49
Cdc13 plays dual roles in telomere length maintenance through its interaction with telomerase and Stn1-Ten1 complexes.Citation34,39 In Li et al., 2009, Cdk1-dependent phosphorylation of Cdc13 during late S phase was found to be essential for recruiting telomerase to the telomeres to promote telomere elongation.Citation27 Interestingly, it also suggested that the recruitment of Est1 and Stn1 to telomeres overlapped during cell cycle. In our earlier work, we reported that Cdk1-dependent phosphorylations of Stn1 acts as the molecular switch that promotes the association of Cdc13 with Stn1-Ten1 rather than telomerase.Citation28 We showed that Cdk1-dependent phosphorylation of Stn1 is sequential to Cdc13 phosphorylation leading to the formation of a stable CST complex that inhibits telomerase at telomeres in late S/G2 phase of cell cycle. Although Cdk1 phosphorylates both Cdc13 and Stn1 at successive cell cycle stages, it is unclear how a single kinase temporally regulates different proteins of the same complex that elicits opposing functions at the telomere.
Fluctuating levels of different cyclins that confer substrate specificity to cyclin-CDK (cyclin-dependent kinase) complexes in combination with gradually increasing levels of CDK activity during cell cycle progression trigger irreversible transitions from G1 to S, entry into mitosis and metaphase to anaphase during cell cycle progression.Citation50-52 In budding yeast, the cyclins that partner with the sole cyclin-dependent kinase, Cdk1, in different phases of the cell cycle are: Cln1, Cln2 and Cln3 in G1 that also trigger G1/S transition; Clb5 and Clb6 in driving S phase; Clb3 and Clb4 in G2/M; and Clb1 and Clb2 in M phase (major cyclin in each phase underscored, Figure S1A, reviewed in Citation53). In addition to the increasing intrinsic selectivity of the CDK active site, different cyclins control the CDK substrate specificity through the substrate docking site on the cyclin subunit.Citation50 Studies have identified different consensus motifs/sites, which determine the efficiency of different cyclin-Cdk1 complexes in phosphorylating their substrates.Citation50,53-55 The early phase cyclins contain hydrophobic patch that interacts with a conserved motif Clb5- or Clb3- specific docking motif, R-X-L, on the substrate.Citation50 This enables even low levels of CDK activity, typically observed during early phases of the cell cycle, to phosphorylate S phase substrates at its optimal consensus motif (S/T-P-X-K/R) with high efficiency.Citation56-59 On the other hand, the lack of Clb2-specific docking site together with suboptimal consensus motif (S/T-P) prevent the efficient phosphorylation of other substrates until CDK activity reaches a much higher threshold, essentially at the later stage of cell cycle. This allows a mechanism in which substrates with optimal sites and suboptimal sites are phosphorylated during the early and late cell cycle respectively.Citation57
Here, we show that the resolution of the molecular switch between telomerase-extendable and CST-unextendable states is driven by different cyclins that confer substrate specificity to Cdk1 at different stages of the cell cycle supplemented by increasing Cdk1 activity. We find that the S phase cyclins which are responsible for the progression of DNA replication, target telomeric protein, Cdc13, and positively regulates telomerase while the mitotic cyclins target Stn1 for phosphorylation by Cdk1. Our results suggest a mechanism, that allows a single kinase to control opposing functions of telomere maintenance and highlights a previously unappreciated role of cyclins in telomere replication.
Results
Loss of S Phase cyclins result in telomere shortening that recapitulates the telomere phenotype of Cdc13 phosphorylation mutant cdc13-T308A
Since the phosphorylation of Cdc13 occurs during S-phase, we hypothesized that Cdk1-dependent phosphorylation of Cdc13 is facilitated by the S Phase cyclins, Clb5 and Clb6 (Figure S1A). Additionally, Cdc13 contains a well conserved RXL domain downstream of the conserved Cdk1 phosphorylation site (T-P-E-R), characteristics that define early cyclin specificity and optimal CDK phosphorylation site (Figure S1B). Loss of Cdk1-dependent phosphorylation of (T308) in Cdc13 is known to result in telomere shortening.Citation27 To evaluate the role of S Phase cyclins in Cdc13 phosphorylation and its subsequent effect on telomere maintenance, we engineered yeast strains that lack Clb5, Clb6 or Clb5/Clb6, and evaluated their telomere length. A diploid yeast strain in A364a genetic background, heterozygous for Δclb5, Δclb6 and cdc13-T308A, was sporulated and dissected to obtain different resultant genotype combinations including: Wild type (WT); single mutants Δclb5, Δclb6, cdc13T308A; double mutants Δclb5 Δclb6, cdc13T308A Δclb5, cdc13T308A Δclb6 and triple mutant that lack both S Phase cyclins (Δclb5, Δclb6) and harbors the cdc13 phosphorylation mutant cdc13T308A. Yeast colonies derived from the individual spores harboring each of the above mentioned genotypes were grown for ∼100 generation at 30°C on YPD plates. To assess the telomere length, Teloblot (Southern Blot using a telomere specific probe) was performed on genomic DNA (gDNA) prepared from the yeast colonies. While yeast harboring single deletion of either Δclb5 or Δclb6 did not show any obvious change in the telomere phenotype, yeast harboring double deletion of both the S phase cyclins, Δclb5 Δclb6, resulted in shorter telomeres (). Since cyclins are largely redundant in their functions, the loss of individual S phase cyclins do not alter cell cycle regulations.Citation60 Due to redundancy, the lack of a difference in telomere phenotype between WT and single mutants harboring either Δclb5 or Δclb6 was anticipated. Hence a combined deletion of both Clb5 and Clb6, thereby obstructing the specific regulations of the S Phase cyclins, is necessary to gauge the role of these cyclins in telomere replication. Interestingly, loss of both Clb5 and Clb6 results in shorter telomeres as shown in . Intriguingly, the telomere shortening observed in strains lacking both S phase cyclins is similar to the short telomeres observed in the Cdk1-dependent phosphorylation mutant of Cdc13, cdc13T308A ( compared with ). Furthermore, the telomere phenotype of double mutants combining loss of Clb5 or/and Clb6 with cdc13T308A does not result in an additive telomere shortening. These results suggest that the function of the S Phase cyclins at the telomeres in vivo is largely mediated through the Cdk1-dependent phosphorylation of Cdc13.
Figure 1. (S)Phase cyclins facilitate Cdk1-dependent phosphorylation of Cdc13 in vivo. (A) Measuring telomere length using teloblot: Loss of individual (S)phase cyclins show no change in telomere length (Compare lanes 1,2,17,18 to 3–6). Combined deletion of (S) phase cyclins, Δclb5Δclb6, result in shortened telomeres identical to the telomere phenotype observed in Cdc13 phosphorylation mutant, cdc13-T308A (Compare lanes 13,14 and 7,8 to 1,2,17,18). Additionally, cdc13-T308A along with Δclb5 or Δclb6 does not alter the short telomere phenotype observed in cdc13-T308A (Compare lanes 7,8 and 9–12). (B) Cell cycle dependent phosphorylation status of Cdc13(T308) in Δclb5Δclb6 vs WT: Haploid yeast strains carrying 3xFlag tagged Cdc13 with either WT (Top Panel) or Δclb5Δclb6 (bottom panel) was arrested in G1 using α-factor and released to progress through cell cycle collecting pellets every 15 min. Immunoprecipitation of Cdc13–3xFlag with anti-Flag in cell cycle synchronized yeast cells shows that there is a 30 min delay in Cdc13 phosphorylation in the strain lacking Clb5/6 compared with the WT. (C) FACS analysis indicative of the cell cycle progression after α-factor synchronization for strains used in ‘(B)’ shows that the onset of (S) phase is delayed by 30 min for Δclb5Δclb6 compared with WT.
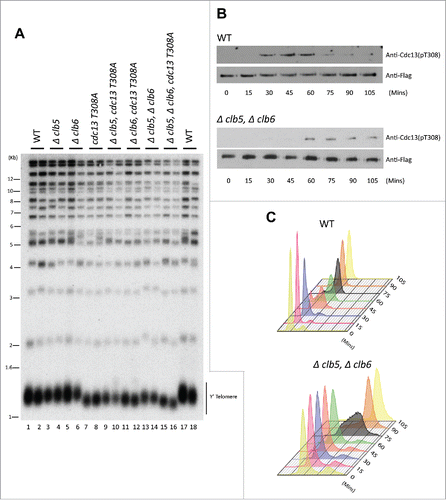
Clb5 and Clb6 control the timing of Cdk1-dependent phosphorylation of Cdc13 at T308.
In line with the idea that the role of S Phase cyclins in telomere maintenance is through the phosphorylation of Cdc13 by Cdk1, we determined how Clb5 and Clb6 affects the timing of Cdc13 phosphorylation at T308 in vivo. For this, we grew haploid yeast cultures of WT strains or yeast lacking Clb5/6 cyclins that were arrested at G1 using α-factor and then released to progress through cell cycle. Whole cell lysates prepared from synchronous yeast cultures collected every 15 min, were used for immunoprecipitation using anti-Flag antibody to pull down 3xFlag-tagged Cdc13. As seen in , the lack of Clb5 and Clb6 delays the timing of Cdc13 phosphorylation by 30 min compared with the WT, as indicated by the Cdc13-T308 phospho-specific antibody. This coincides with the delay in the onset of S Phase observed in Δclb5 Δclb6 compared with WT, as evidenced by the analysis of cellular DNA content using Fluorescence Activated Cell Sorting (FACS) in . The delay in Cdc13 phosphorylation caused by loss of S phase cyclins is consistent with the delay in the S phase onset in these strains. This is analogous to the delayed but uninhibited progression of S phase in cells lacking Clb5 and Clb6.Citation60 The mitotic cyclins compensate for the loss of S phase cyclins in their regulatory functions.Citation60-62 This is consistent with the phosphorylation of Cdc13 detected, though delayed, in cells lacking S phase cyclins. It is possible that the efficiency of Cdc13 phosphorylation in the absence of S phase cyclins is not optimal in vivo. These in vivo results, taken together, are consistent with the notion that S phase cyclins target Cdc13 for phosphorylation by Cdk1, thereby controlling the timing of Cdc13 phosphorylation and influencing the onset of telomerase-dependent telomere elongation.
S Phase Cyclins are essential for the recruitment of Cdc13 and Est1 but not Stn1
While telomerase is expressed and enzymatically active throughout the cell cycle, its function of telomere elongation is restricted to late S to G2 phases primarily caused by its recruitment to the telomeres.Citation63 Telomerase recruitment is facilitated by the interaction between the telomerase subunit Est1 and Cdc13.Citation34,38 Cdk1-depedent phosphorylation of Cdc13 at T308 during S phase is necessary for the efficient association of Cdc13 and Est1, and subsequent recruitment of telomerase to telomeres, to promote telomere elongation. Since the lack of S phase cyclins affects the timing of Cdk1-dependent phosphorylation of Cdc13 and results in telomere shortening, we set out to investigate whether telomere shortening was a consequence of the inability of the S phase cyclins to efficiently facilitate Cdc13 phosphorylation in vivo, thereby compromising the interaction between Cdc13 and Est1 and, subsequently, the recruitment of telomerase to the telomeres. Loss of Cdk1-dependent phosphorylation of Cdc13 at T308 reduced the recruitment of telomerase, but not of Stn1, to telomeres.Citation27 So, we hypothesized that loss of S phase Cdk1-partners will have a similar effect on the recruitment of CST complex components and telomerase to telomere. To test this, we performed Chromatin Immunoprecipitation (ChIP) assay on cell cycle synchronized as well as asynchronous WT and Δclb5 Δclb6 cells. As shown in , loss of the S phase cyclins not only delays but also dramatically compromises the recruitment of Cdc13 to telomeres. The 30-min delay in the recruitment of Cdc13 observed in Δclb5 Δclb6 strains compared with WT correlates with the delay in the onset of S phase in Δclb5 Δclb6 strains as evidenced by their FACS profile (). These results taken together with the delay seen in the timing of Cdc13 phosphorylation in suggest that the S phase cyclins facilitate Cdk1-dependent phosphorylation of Cdc13 in vivo, leading to the recruitment of Cdc13 to telomeres. Consequently, the recruitment of Est1 is also delayed and compromised in Δclb5 Δclb6 ( and FACS profile in ). This confirms that the telomere shortening observed in Δclb5 Δclb6 is due to reduced recruitment of telomerase to the telomeres. While Cdc13 and Est1 recruitment to telomeres is compromised, ChIP assay performed on synchronous yeast cultures indicate that the recruitment of Stn1 to telomeres, despite being delayed is otherwise unchanged by the lack of Clb5 and Clb6 ( and FACS profile in ). This suggests that Cdk1-dependent phosphorylation of Stn1, a prerequisite for Stn1s recruitment to telomeres,Citation28 is likely not facilitated by Clb5 and Clb6. This may be sufficient to tilt the balance toward the Cdc13-Stn1 interaction over Cdc13s interaction with telomerase, leading to the formation of a stable CST complex that negatively regulates telomerase-dependent telomere elongation. The results from synchronous cultures reflect the ChIP data observed in asynchronous cells (Fig. S1C) which indicate that loss of S phase cyclins compromises recruitment of Cdc13 and Est1 but not Stn1 to telomeres. Collectively, these results suggest that the S phase cyclins specifically target Cdc13 but not Stn1 for phosphorylation by Cdk1 in vivo.
Figure 2. Clb5 and Clb6 are essential for the recruitment of Cdc13 and Est1 but not Stn1. Haploid yeast strains harboring 13xMyc tagged Cdc13, Stn1 or Est1 in a genetic background of WT or Δclb5Δclb6 was arrested in G1 using α-factor and released to progress through cell cycle collecting 15 min time points for the ChIP assay. ChIP was performed to evaluate the recruitment of Cdc13, Stn1 or Est1 in cell cycle synchronized WT vs Δclb5Δclb6 yeast cultures. (A) ChIP assay indicates that the cell cycle dependent recruitment of Cdc13 to telomeres is delayed and compromised in synchronous yeast cultures harboring Δclb5Δclb6 compared with WT. (B) FACS profile indicative of the cell cycle progression of synchronous yeast cultures used for the ChIP assay in ‘A’ - the onset of (S)phase is delayed by 30 min in Δclb5Δclb6 compared with WT. (C) ChIP assay indicates that the cell cycle dependent recruitment of Est1 to telomeres is delayed and compromised in cell cycle synchronized yeast cultures harboring Δclb5Δclb6 compared with WT. (D) FACS profile indicative of the cell cycle progression of synchronous yeast cultures used for the ChIP assay in ‘C’. (E) ChIP assay indicates that the cell cycle dependent recruitment of Stn1 to telomeres is delayed but not compromised in synchronous yeast cultures harboring Δclb5Δclb6 compared with WT. (F) FACS profiles indicative of cell cycle progression of synchronous yeast cultures used for the ChIP assay in ‘E’.
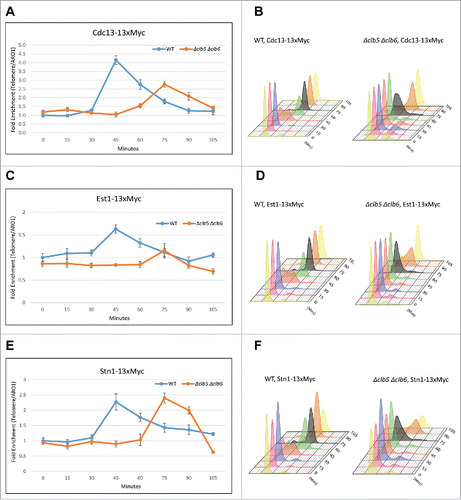
Loss of G2/M or M phase cyclins do not alter telomere phenotype and the timing of Stn1 phosphorylations by Cdk1.
We reported in our earlier work that the loss of Stn1 phosphorylations results in increased telomere length since telomerase-dependent telomere elongation is uninhibited in the absence of a stable CST complex, a consequence of Stn1 phosphorylations and its recruitment to telomeres.Citation28 Given that Stn1 possesses no optimal CDK motifs around its phosphorylation sites and that the S phase cyclins do not affect Stn1 recruitment to the telomeres, it suggests that Stn1 is unlikely to be a target of Clb5 and Clb6. Furthermore, the Cdk1-dependent phosphorylations of Stn1 sequentially follows that of Cdc13.Citation28 Therefore, we hypothesized that the mitotic cyclins could likely be facilitating the Cdk1-dependent phosphorylations of Stn1 thereby negatively influencing telomere elongation. To test this, we first wanted to evaluate the effect of the G2/M cyclins on telomere phenotype. For this, we engineered diploid yeast strains heterozygous for Δclb3 Δclb4, sporulated and dissected them to obtain single mutants, Δclb3 or Δclb4, and double deletion mutants lacking both Clb3 and Clb4. Teloblot was then performed as described above. As seen in , teloblot shows that loss of G2/M phase cyclins, neither individually nor in combination, affect telomere length compared with WT. Since the loss of G2/M cyclins is compensated by the late phase cyclins, Clb1 or Clb2, we wanted to test whether the lack Clb1 or Clb2 caused an increase in telomere length as observed in stn1-T223A,S250A.Citation28 As shown in , loss of either Clb1 or Clb2, respectively, does not affect telomere length either. Given the redundancy of the mitotic cyclins, these results are not surprising. However, the experimental limitations posed a challenge to evaluate the effect of a combined deletion of Clb1 and Clb2, as Δclb1Δclb2 is lethal.Citation61 Furthermore, Clb2 being the most important of the B-type cyclin required for survival, (and clb4, the least important), combining Δclb2 with deletions of any of the other G2/M or M phase cyclins (Δclb3, Δclb4, or Δclb1) would render the yeast inviable and hence could not be investigated in our study.Citation60,61 Therefore, while it can be concluded that the loss of G2/M or M phase cyclins alone does not alter telomere length homeostasis, the possibility that the mitotic cyclins specifically target Stn1 for phosphorylation by Cdk1 in vivo cannot be ruled out.
Figure 3. G2/(M)or M-phase cyclins do not alter telomere phenotype or the timing of Cdk1-dependent phosphorylation of Stn1 during cell cycle. (A) The first panel shows that telomere length measured for each of the genotypes, Δclb3, Δclb4, double mutant Δclb3Δclb4 or WT, indicates that the telomere length is unaltered in mutants lacking Clb3 and/or Clb4 compared with WT; the 2nd and 3rd panels show that loss of mitotic cyclins, Clb1 or Clb2 respectively, do not alter telomere length compared with WT. (B) Cdk1 dependent phosphorylation status of Stn1 for sites T223 and S250 were tested in yeast strains harboring deletions of different S- to M-phase cyclins compared with WT. Haploid yeast strains carrying 13xMyc tagged Stn1 with either WT (Top Panel) or Δclb5Δclb6 / Δclb3Δclb4 / Δclb2 (bottom 3 panels) was arrested in G1 using α-factor and released to progress through cell cycle with cultures collected every 15 min. Immunoprecipitation of Stn1–13xMyc with anti-Myc show no obvious difference in the profile of Stn1 phosphorylation detected with phospho-specific antibody Stn1-pS250 in synchronous yeast cultures harboring Δclb3Δclb4 / Δclb2 compared with WT. The phosphorylation of Stn1 in Δclb5Δclb6 coincides with the onset of S phase as shown in the FACS profile in ‘C’. (C) FACS profiles showing the cell cycle progression of the α-factor synchronized yeast cultures in ‘B’ – while loss of Clb5/6 delays the onset of S phase to 75 mins, neither Δclb3/4 nor Δclb2 cause variation to cell cycle progression compared with WT.
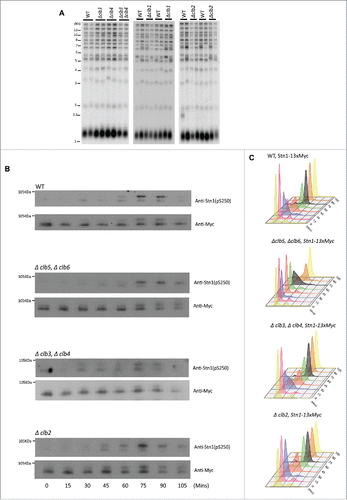
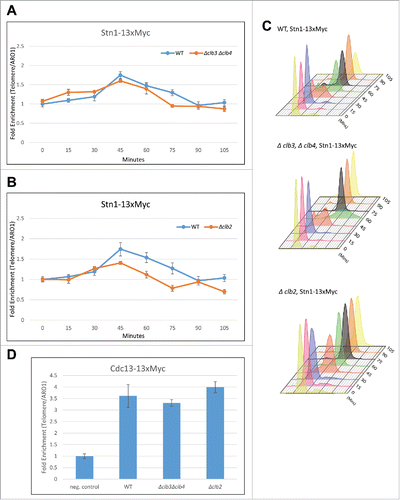
While the S phase cyclins do not affect the level of recruitment of Stn1 to the telomeres, the timing of its recruitment to telomeres is delayed coinciding with the delay in the onset of S phase observed in Δclb5/6 (). So, to test whether the cyclins affected the timing of Stn1 phosphorylation, we performed immunoprecipitation using lysates from cell cycle synchronous yeast cultures. As shown in , the timing of Stn1 phosphorylation coincides with the onset of S phase in synchronous yeast cultures harboring Δclb5Δclb6. This result suggests that the S phase cyclins prevent the early phosphorylation of Stn1, perhaps as a means to prevent the early inhibition of telomerase access to telomeres. This is consistent with the finding that low S phase cyclin-CDK activity prevents early onset of mitosis (reviewed in Citation64). Furthermore, Stn1 is likely a better candidate for phosphorylation by the mitotic cyclins, which promote S phase onset and progression in the absence of Clb5/6. This is consistent with Cdk1-dependent phosphorylations of Stn1 occurring during late S, peaking at G2/M phase and lasting through most of the mitotic phase of the cell cycle in WT, evidenced by the appearance of the slower migrating form of Stn1 as well as Stn1 with phosphorylated serine 250 as probed by S250 phospho-specific antibody in the immunoprecipitation and western blot analysis of WT synchronous cells (, Citation28). To obtain further resolution on this, we tested whether the lack of G2/M or M phase cyclins affected the timing of Stn1 phosphorylation. Intriguingly, we did not observe any obvious difference in the timing of Stn1 phosphorylation harboring either Δclb3Δclb4 or Δclb2 (the major M phase cyclin) (). As shown in our previous work in Liu et al., (2014), the slower migrating band is indicative of Stn1 phosphorylation at both sites T223 and S250. These results are likely due to the redundancy of cyclins in mediating the phosphorylation of Stn1 such that the lack of either G2/M or M phase cyclins does not affect the timing of cell cycle events. This is consistent with the lack of an obvious difference in cell cycle progression besides being mildly sluggish in G2/M to M for Δclb3Δclb4 or Δclb2 compared with WT as indicated by their corresponding FACS profiles in . Interestingly, we also did not observe any difference in the timing of Stn1 phosphorylation in cdc13-T308A background (Figure S2). This suggests that Stn1 phosphorylation is independent of the timing of Cdc13s phosphorylation or dephosphorylation, and the onus in determining the timing and efficiency of Stn1 phosphorylation is perhaps on the cyclins that target Stn1 and the threshold of Cdk1 activity.
Clb2 is responsible for the recruitment of Stn1 to telomeres
Cdk1-dependent phosphorylation of Stn1 at T223 and S250 is necessary for the recruitment of Stn1 to telomeres.Citation28 While the deletion of G2/M or M phase cyclins did not affect the timing of Stn1 phosphorylation, we wanted to test whether the efficiency of the Cdk1-dependent phosphorylation of Stn1 was affected in yeast strains with deletion in G2/M and M phase cyclins. One of the readouts to test this is to analyze the recruitment of Stn1 to telomeres in Δclb3Δclb4 or Δclb2 compared with WT. Therefore, we performed ChIP assay on synchronous cells harboring either Δclb3Δclb4 or Δclb2 and compared it to WT. As shown in , in yeast strains lacking G2/M cyclins, Δclb3Δclb4, Stn1 is recruited to telomeres in a cell-cycle-dependent manner, similar to WT. The cell cycle profile obtained from FACS analysis indicates that the timing of Stn1 recruitment to telomeres is also unaltered and occurs during late S to G2 phases of the cell cycle in Δclb3Δclb4 and WT (). This suggests that the Stn1 is an unlikely substrate for phosphorylation by Cdk1-Clb3/4. Interestingly, in the ChIP assay performed on synchronous yeast cultures harboring Δclb2 vs WT, we observed that the cell cycle dependent recruitment of Stn1 to telomeres is compromised in Δclb2 compared with WT while the FACS analysis indicate no difference in cell cycle progression profile between Δclb2 and WT cells (). These results, taken together with the fact that Stn1 only consists of suboptimal CDK sites (S/T-P) (likely targeted by late phase cyclins), support the idea that Stn1 is a candidate for phosphorylation by Cdk1-Clb2. To further understand whether Clb2 specifically targets Stn1 and to analyze the role of G2/M and M phase cyclins in telomere replication, we tested how they affected the recruitment of the other CST component, Cdc13. Since the Cdc13 phosphorylation is dependent on S phase cyclins, we hypothesized that the recruitment of Cdc13 to telomeres would be unaffected by mitotic cyclins. As expected ChIP assay performed on asynchronous cultures of haploid yeast strains harboring Δclb3Δclb4, Δclb2 or WT, indicates that the recruitment of Cdc13 to telomeres is not different in Δclb3Δclb4 or Δclb2 cells compared with WT (). Collectively, these results strengthen the proposition that the mitotic cyclin, Clb2, specifically targets and regulates Stn1 but not Cdc13.
Figure 4. Clb2 but not Clb3/4 is responsible for the recruitment of Stn1 to telomeres in vivo. (A) ChIP assay indicates that the cell cycle dependent recruitment of Stn1 to telomeres shows small or no change in synchronous yeast cultures harboring Δclb3Δclb4 compared with WT. (B) ChIP assay indicates that the cell cycle dependent recruitment of Stn1 to telomeres is compromised in synchronous yeast cultures lacking Clb2 compared with WT. (C) FACS profiles indicative of cell cycle progression in synchronous cultures used in ‘A’ and ‘B’. (D) ChIP assay in asynchronous yeast cultures with 13xMyc tagged Cdc13 indicates that the recruitment of Cdc13 to telomeres is unaffected in Δclb3Δclb4 or Δclb2 compared with WT.
Clb2-Cdk1 phosphorylates Stn1 more efficiently in vitro compared with other Cyclin-Cdk1 complexes
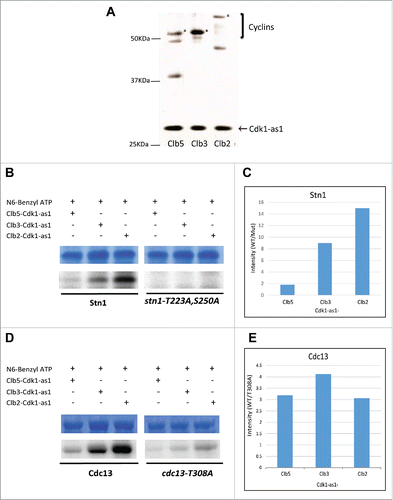
Thus far, in vivo results suggest that the Cdk1-dependent phosphorylations of Cdc13 and Stn1 are facilitated by Clb5/Clb6 and Clb2 respectively. To further validate these results, we performed in vitro kinase assay, in which different cyclin-Cdk1-as1 complexes were incubated with 6xHis-tagged WT or phosphorylation mutants of Stn1(M) and Cdc13. The representative cyclins of each cell cycle phase – Clb5 for S Phase, Clb3 for G2/M and Clb2 for M Phase, were TAP (Tandem Affinity Purification) tagged in separate yeast strains harboring Cdk1-as1, an analog of Cdk1 that specifically uses bulky N6-Benzyl-ATP source for its kinase activity. The TAP tag purification of cyclin-Cdk1-as1 complexes were performed using asynchronous yeast cultures ( and Fig. S3).Citation65,66 After normalizing for equal amounts of different cyclin-cdk1-as1 complexes using silver stained-SDS-resolved gels, the kinase activity of the Cyclin-Cdk1-as1 complexes was validated through in vitro kinase assay performed using Histone H1 as an optimal model substrate for Cdk1 (Fig. S4). Consistent with previous reports, quantitative analysis of Histone H1 phosphorylation by different Cyclin-Cdk1 complexes showed that Histone H1 is phosphorylated more efficiently by Clb2-Cdk1 compared with Clb3-Cdk1 and Clb5-Cdk1 (Figure S4, Citation53). Hence, we tested the phosphorylation efficiency of the cyclin-Cdk1 complexes on Stn1 and Cdc13, using the bacterially expressed recombinant 6xHis-tagged WT or phosphorylation mutants of Stn1(M) and Cdc13 (Figure S5 and Figure S6A). As shown in , the in vitro kinase assay indicates that the specificity of mitotic Clb2-Cdk1 for wild-type Stn1 is much greater compared with Clb3-Cdk1 or Clb5-Cdk1 complex. In contrast, the phosphorylation was completely abolished when His-tagged stn1-T223A,S250A was used as the substrate. This is consistent with the in vivo ChIP data suggesting that the mitotic cyclin Clb2 facilitates the phosphorylation of Stn1 with much greater efficiency compared with the earlier phase cyclins Clb3 and Clb5. Intriguingly, the in vitro kinase assay using Cdc13–6xHis (WT and T308A mutant) as the substrate shows no significant specificity for any particular Cdk1-cyclin complex (). The lack of specificity for Cdc13 by the cyclin-Cdk1 complexes in vitro can be attributed to the added complexity in Cdc13 regulation in vivo including multiple potential phosphorylation sites in Cdc13.Citation67 To understand if other phosphorylation sites on Cdc13 contributed to its phosphorylation in vitro, we performed the assay using cdc13–7A as the substrate, in which all putative Cdk1 phosphorylation sites had been mutated to alanine (Figure S6). Here, we noticed that the ratio of phosphorylation efficiency for Cdc13 wild type over phosphorylation mutant (cdc13–7A), was greater for Clb3-Cdk1 compared with Clb5 or Clb2 (Figure S6). Furthermore, a low level of phosphorylation was observed even when all putative Cdk1 phosphorylation sites were mutated. This suggests that there are multiple phosphorylation sites in Cdc13 targeted by Cdk1 which perhaps mildly skews the specificity toward Clb3-Cdk1. Furthermore, the docking sites and consensus motifs for Clb5- and Clb3- specific substrates are similar in that Clb3, like Clb5, also targets Cdk1 substrates through the RXL hydrophobic patch interactions in addition to the optimal consensus motif of S/T-P-X-K/R, which Cdc13 possesses.Citation53 In addition to their redundancy, Clb5 and Clb3 have significantly overlapping temporal windows in cell cycle allowing these cyclins to act on similar targets during different phases of the cell cycle.Citation61,62 In vitro kinase assay results suggest that Cdc13 can be phosphorylated efficiently by all 3 cyclin-Cdk1 complexes. However, only the S phase cyclins influence the phosphorylation of Cdc13 in vivo as evidenced by the delay and compromise in the recruitment of Cdc13 to telomeres in yeast strains lacking S phase cyclins. In the absence of S phase cyclins, the G2/M cyclins can compensate, resulting a delay in Cdc13 phosphorylation in yeast harboring Δclb5Δclb6. So, while Cdc13's phosphorylation can be compensated by G2/M and M phase cyclins in vitro, it is likely antagonized by phosphatase in vivo which actively reverses the Cdk1-dependent phosphorylation during these phases.Citation68 Taken together, our results from in vivo and in vitro experiments suggest that the sequential phosphorylation of Cdc13 and Stn1 is facilitated by S phase and mitotic cyclins, respectively.
Figure 5. Clb2 facilitates the phosphorylation of Stn1 in vitro while Cdc13 phosphorylation lacks significant specificity for any particular Cyclin-Cdk1 complex in vitro. (A) TAP-tagged Cyclins were purified in yeast strains harboring Cdk1-as1 allele. The silver stained gel shows the different Cyclin-Cdk1-as1 complexes purified through TAP-tag purification. *, Clb5/Clb3/Clb2. (B) In vitro Kinase assay analyzing the differential phosphorylation efficiency of Stn1(M) by different Cyclin-Cdk1-as1 complexes. The top panel shows the coomassie blue stained input, purified 6xHis tagged recombinant Stn1(M), WT and stn1-T223A, S250A. The bottom panel shows the corresponding autoradiograph from the kinase assay. (C) Quantification of the corresponding phosphorylation efficiency (WT/mutant) obtained from the autoradiograph over input in ‘B’ shows that Clb2-Cdk1-as1 phosphorylates Stn1(M) with greatest efficiency followed by Clb3-Cdk1-as1 and finally Clb5-Cdk1-as1. (D) In vitro Kinase assay analyzing the differential phosphorylation efficiency of Cdc13 by different Cyclin-Cdk1-as1 complexes. The top panel shows the coomassie blue stained input, purified 6xHis tagged recombinant Cdc13, WT and mutant (T308A). The bottom panel shows the corresponding autoradiograph for the kinase assay. (E) Quantification of the corresponding phosphorylation efficiency (WT/mutant) obtained from the autoradiograph over input in (D) shows no significant specificity of any particular Cdk1-cyclin for phosphorylation of Cdc13 in vitro.
Discussion
In budding yeast, Cdk1 is the master regulator of telomere replication. However, the underlying molecular mechanisms and targets of Cdk1-dependent phosphorylation remained largely uncharacterized. Li et al., (2009) reported that Cdk1-dependent phosphorylation of Cdc13 at T308, promotes its interaction with Est1 and the recruitment of telomerase to telomeres, thereby promoting telomere elongation.Citation27 In our earlier work, we showed that Cdk1 also phosphorylates Stn1, resulting in the formation of stable CST complex that inhibits telomerase at telomeres.Citation28 While Cdc13 and Stn1 are both phosphorylated by Cdk1, it was intriguing to find their phosphorylations to be sequential during cell cycle progression and exerting opposing outcomes in telomerase-dependent telomere elongation. In this study, we sought to obtain resolution of the dynamic molecular events that orchestrate telomere replication by studying the sequential phosphorylation of Cdc13 and Stn1 by Cdk1. We focused on the cyclins that supplement the intrinsic selectivity of Cdk1 and control the timing of phosphorylation of various Cdk1 substrates during each phase of the cell cycle.Citation50,53 We have characterized how Cdk1-dependent phosphorylation of Cdc13 and Stn1 are temporally coordinated by different cyclins that confer substrate specificity to Cdk1 through successive stages of the cell cycle.
Phase-specific, B-type cyclins are largely redundant in function and are non-essential genes.Citation60,62,69,69,70 While simultaneous deletions of Clb5 Clb3 Clb4, Clb1 Clb2, Clb2 Clb3 or Clb1–4 are inviable, strains lacking Clb1 Clb3 Clb4, Clb5 Clb2 and Clb2 Clb4 are viable.Citation60,71,72 Hence it is interesting to note that, in our results, despite the redundancy of cyclins, the loss of S phase-specific cyclins but not mitotic-specific cyclins substantially impair telomere maintenance, suggesting an indispensable role for Clb5 and Clb6 in telomere maintenance (). Because experimental limitations make it challenging to evaluate the effect of Clb1–4 on telomere phenotype, we cannot rule out the possibility that G2/M and M phase cyclins, together or independently, being essential factors in regulating telomere homeostasis.
While loss of Clb5 and Clb6 does not cause lethality, it delays the onset of S Phase.Citation69-71 This is consistent with our results in which we observe a delay in telomere replication evidenced by a delay in the recruitment of telomeric proteins in Δclb5Δclb6 compared with wild-type (). The Cln and Clb1–4 cyclins are known to eventually compensate for the loss of S phase cyclins, particularly by initiating S phase and activating some of the replication origins.Citation60,70,71,73 Interestingly, the delay in the onset of S phase correlates with the delay in Cdk1-dependent phosphorylation of Cdc13 (). Furthermore, the loss of S phase cyclins compromises the recruitment of Cdc13 to telomeres and thereby, telomerase to telomeres, resulting in short telomeres (). While Li et al., (2009) reported that the recruitment of Cdc13 in cdc13-T308A is not that much different than in WT, the dramatic reduction in recruitment of Cdc13 observed in Δclb5Δclb6 strains can be attributed to potential multisite phosphorylation of Cdc13, which may control its recruitment to telomeres with greater effect.Citation74 Furthermore, we cannot rule out the possibility that the loss of S phase cyclins influences other targets in the telomere replication machinery which has an additive effect on Cdc13 recruitment to telomeres. Additionally, since the recruitment of Cdc13 does not always have to correlate with that of telomerase, it is possible that Cdk1, independently, influences the activation, recruitment and assembly of telomerase components to telomeres (reviewed in Citation75). It will be important to further characterize these potential Cdk1-dependent phosphorylation events in subsequent studies. Taken together, our results suggest that Clb1–4 are not sufficient to trigger and control the progression of telomere replication in the absence of Clb5/6 emphasizing a previously unidentified role of S phase cyclins in telomere replication. The shorter telomeres () and compromised recruitment of Est1 but not Stn1 to telomeres in Δclb5Δclb6 () resembles cdc13T308A mutants, implying that the function of Clb5/6 at the telomere is through the regulation of Cdc13. This aligns with the existing model in which Cdk1-dependent phosphorylation of Cdc13 is one of the initial steps in telomere elongation as it brings telomerase to the telomeres to carry out its function.Citation27 Considering that the recruitment of Cdc13 and Stn1 is dependent on their phosphorylation, our results strongly suggest that the S phase cyclins are responsible for the phosphorylation of Cdc13 but not for Stn1 phosphorylation.
Stn1 does not possess the optimal consensus motifs that define early targets of Cdk1 during S phase. This suggests that CDK activity is required to reach a much higher threshold before activating suboptimal targets like Stn1. This is consistent with the observation that Stn1 phosphorylation occurs during late S phase, peaking at G2 and lasts until the exit from mitosis (Citation28 and ). The timing of Stn1 phosphorylation is not affected in strains lacking any of the mitotic cyclins although the lack of S phase cyclins advances Stn1 phosphorylation to the onset of S phase (). This indicates that – (a) S phase cyclins prevent early onset of Cdk1-dependent phosphorylation of Stn1 and (b) mitotic cyclins that compensate for lack of Clb5/6 in S phase progression, are activated earlier in Δclb5Δclb6 strains and therefore causing Stn1 to be phosphorylated earlier. Furthermore, the redundancy of the individual mitotic cyclins could explain why there is no obvious difference in the timing and progression of Stn1 phosphorylation in strains lacking G2/M or M phase cyclins. In , it can be observed that phosphorylation of Stn1 is comparatively less efficient in Clb3/Clb4 deletion mutant, although there is no effect on the recruitment of Stn1 to telomere (). It is possible that loss of Clb3/Clb4 may activate a positive regulatory mechanism that stimulate the recruitment of Stn1 to the telomere. Furthermore, Stn1 expression levels in vivo are much higher compared with that of Cdc13. So, it is likely that the mild difference in the phosphorylation efficiency does not lead to significant changes in recruitment to telomeres and inhibition of telomerase. Furthermore, Clb2-Cdk1, which is also shown to phosphorylate Stn1 with highest efficiency in vitro (), can compensate for loss of Clb3/4 in vivo. This is evidenced by results in in which the loss of G2/M cyclins causes small or no change in the recruitment of Stn1, but loss of Clb2 substantially reduces Stn1 recruitment to telomeres. The lack of telomere lengthening phenotype in strains lacking mitotic cyclins () despite compromised Stn1 recruitment in Δclb2 could be due to 2 factors – (a) redundancy of the cyclins and (b) lack of Clb2 possibly inhibiting a positive regulatory mechanism of telomerase, eventually resulting in WT length of the telomere in Δclb2. Taken together with the in vitro kinase assay in which Stn1 shows high level of specificity to phosphorylation by Clb2-Cdk1 (), our results suggest that Stn1 is a target of Cdk1-Clb2 complex. This is consistent with the existing model that Cdk1-dependent phosphorylation of Stn1 promotes the recruitment of Stn1-Ten1 to telomeres to form a stable CST complex that inhibits telomerase at telomeres after the telomere is elongated. Thus, Stn1 phosphorylation serves as a late “switch” in cell cycle that sequentially follows telomere elongation. Considering that Stn1 also interacts with Pol12 of the DNA polymerase α complex, which is also a Clb2-specific substrate, a late switch based on Stn1 phosphorylation is well positioned to coordinate C-strand fill-in Citation53,76-78
To supplement cyclin specificity, CDK activity increases as cell cycle progresses triggering specific molecular events at every activity threshold - while the low activity of CDK in early phases of the cell cycle is compensated by the optimal conserved motif and docking site on the substrates, later phase targets are phosphorylated only in the presence of high levels of CDK activity.Citation53,56,57,79,80 Furthermore, Cdk1 activity threshold acts as a regulatory mechanism used to prevent licensing and firing of replication origins more than once during a single cell division cycle thereby preventing re-replication of cellular DNA content - the low CDK activity enables DNA replication while higher threshold of CDK activity promotes mitosis (reviewed in Citation81). Cdc13 can be considered an optimal substrate of Cdk1 as it possesses a well conserved consensus motif (S/T-P-X-R) along with the RXL domain that specifies Clb5 or Clb3 targets and is phosphorylated in vivo during S phase of the cell cycle.Citation50,53 Besides parameters associated with CDK activity and cyclin specificity, phosphatases that counteract Cdk1 by dephosphorylating its substrates have a profound role in controlling the timing and intensity of Cdk1-dependent phosphorylation in vivo.Citation80,82 Cdc14, the main phosphatase that offsets Cdk1 activity in vivo, can dephosphorylate early substrates even in the presence of high CDK activity. However, later substrates require higher Cdc14 activity.Citation80,82 The ordered dephosphorylation of Cdk1 targets by Cdc14 is an important mechanism through which cell cycle progression is controlled. These complexities in regulation of CDK activity in vivo could explain the results seen with the in vitro kinase assay which shows little specificity to a single Cyclin-Cdk1 in phosphorylating Cdc13 (). Furthermore, this is not completely surprising given the significant overlap in the common motifs that define substrate specificity for Clb5 and Clb3.Citation53 However, Figure S6 shows that Clb3-Cdk1 phosphorylates Cdc13, WT over 7A (Cdc13 in which all its 7 Cdk1 phosphorylation sites are mutated to Alanine), with mildly greater efficiency in vitro compared with other cyclin-Cdk1 complexes. This suggests that Cdc13 is a target of multisite phosphorylation Citation67 and while Clb5/6 is responsible for triggering the Cdk1-dependent phosphorylation of Cdc13 during S phase, it is possible that intensifying Cdk1 activity and Clb3-Cdk1 potentially maintains the phosphorylation on Cdc13 which is simultaneously counteracted by Cdc14-dependent dephosphorylation during G2/M and M phase of the cell cycle. This way, there is greater emphasis on the negative regulation of telomerase, such that replication and elongation of telomeres do not carry on uninhibited and is indeed restricted to once per cell cycle.
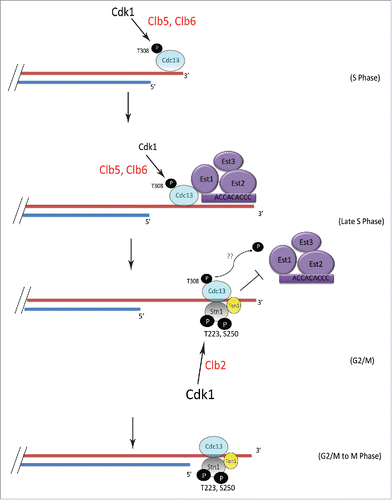
Taken together results from this study, we propose a model () in which S phase cyclins facilitate Cdk1 phosphorylation of Cdc13 at T308 initiating the process of telomere elongation as Cdc13 phosphorylation promotes the recruitment of telomerase to telomeres. It is possible that during the process of telomere elongation Clb3-Cdk1 maintains Cdc13 phosphorylation during G2/M while phosphatase counteracts intensifying CDK activity. As the cells gradually transition from S to G2 and mitosis, Cdk1-dependent phosphorylation of Stn1 at T223, S250 occurs sequentially to that of Cdc13, aided by mitotic cyclins. This stabilizes Stn1 at the telomere, thereby tipping the balance toward a stable Cdc13-Stn1-Ten1 complex and reducing Cdc13-Est1 interaction. While the model indicates the phosphorylations of Cdc13 and Stn1 occur sequentially, the Cdc13-Est1 and Cdc13-Stn1 interactions are considered to be dynamic switches that occur during the late S to G2/M phase of the cell cycle. The phosphorylation of Stn1 is likely coupled to the dephosphorylation of Cdc13, perhaps by Cdc14, promoting the formation of a stable CST complex that inhibits telomerase at telomeres and indeed coordinates C-strand fill in. Preliminary data from our lab suggests a putative role for Cdk1 in regulating DNA Pol α. It would be valuable for future research efforts to shed light on the potential role of the Cyclin-CDK network in the interplay between DNA Pol α and CST components that controls C-Strand synthesis.
Figure 6. Model showing the mechanism of sequential phosphorylations of Cdc13 and Stn1 during cell cycle progression and their role in telomere elongation. S-phase cyclins Clb5/6 initiate Cdk1-dependent phosphorylation of Cdc13 at T308 which promotes the recruitment of Cdc13 and thereby Est1 to telomere, leading to telomere elongation. This is subsequently followed by the sequential phosphorylation of Stn1 at sites T223 and S250 by Cdk1, facilitated by Clb2. The phosphorylation of Cdc13 during G2/M is potentially antagonized in vivo, by phosphatase, perhaps Cdc14, which is involved in dephosphorylating Cdk1 targets (indicated by blue “??”). Thus, the ordered phosphorylation of CST components by different cyclin-Cdk1 complexes, perhaps in combination with phosphatases, regulates telomerase-extendable and CST-unextendable states of the telomere.
While mammalian cyclin-CDK network is much more complex, with many more variants of cyclins and CDK for each phase of the cell cycle, the general principles that govern cell cycle progression and cyclin-CDK specificity are largely conserved from yeast to humans.Citation83 The role of cyclins in telomere replication is especially interesting as it links 2 hallmarks of cancer – immortality, rendered by upregulated telomerase that leads to incessant telomere maintenance, and deregulated cell cycle control caused by upregulation of several Cyclin-CDK complexes.Citation19 Our work sheds light on the synchrony between regulation of replicative immortality and cell cycle control. Recently, Pablociclib, a drug that inhibits Cdk4/6 activity, has been reported to be among the most promising drugs that show improved survival in certain type of breast cancer patients.Citation84,85 With our work unfolding the role of cyclins in telomere replication, it provides an added window of opportunity through which specific cyclin-CDK networks can be targeted such that the incessant telomere lengthening and proliferative potential in cancers can be blocked.
Materials and methods
Saccharomyces cerevisiae was handled using standard techniques. Yeast strains were grown in yeast extract-peptone-dextrose (YPD) media and at optimal temperature of 30°C unless otherwise stated. All yeast strains are in A364a background unless otherwise stated. PCR purified product using Expand™ High Fidelity PCR protocol (Sigma-Aldrich) and the toolbox from Citation86,87 was used to generate templates for epitope tagging of genes or deletion mutants. The yeast strains used in this study are listed in Table S1.
Chromatin immunoprecipitation (ChIP)
Cell cycle synchronization and chromatin immunoprecipitation was done as described previously in.Citation27,28 Briefly, overnight cultures grown at 30°C was diluted to OD600 0.05 and grown to reach an OD600 of 0.3. The cultures were arrested in G1 for 2–2.5 h using α-factor at 0.018µM/ml of culture and then washed and released at 25°C. The cultures collected at 15 min intervals were crosslinked for 30 min with 1% Formaldehyde and neutralized with Glycine following which they were pelleted and used for ChIP assay. For ChIP using asynchronous yeast cultures, overnight yeast culture grown at 30°C was diluted to OD600 0.05 following which the asynchronous cultures were grown to reach confluency of OD600 0.4 to 0.6. Crosslinking with formaldehyde for 30 min was done at 30°C following which the cells were pelleted and used for ChIP experiments.
Chromatin lysates were purified from the yeast pellets using about 1ml ChIP lysis buffer (50mM Hepes pH 7.5, 140mMM NaCl, 1mM EDTA, 10% glycerol, 1% NP-40, 0.1% sodium deoxycholate) and about 1.5ml glass beads. The cells were mechanically disrupted using Mini-Beadbeater (Bio Spec Products) in 4°C. The lysate was then cleared and the chromatin, sheared using Qsonica Q700 Sonicator. The sonication efficiency was confirmed to be in the range of 500bp-1KBp before the equal volume of the chromatin lysates were prepared for pull down using 4µg of anti-Myc 9E10 (QED). The immunoprecipitated proteins with its crosslinked DNA were washed with increasingly stringent buffers, before reverse crosslinking at 65°C. The DNA was then purified using the QIAquick PCR Purification Kit by QIAGEN. Enrichment of the telomere sequence from the purified samples were measured by quantitative polymerase chain reaction (qPCR) using Bio-Rad CFX96 real-time PCR system. Two sets of primer pairs were used in these ChIP experiments: primers for ARO1 amplified a 372 bp sequence in a nontelomeric loci found on chromosome IV and used as the negative control and, primers for TEL which amplified 114bp of a unique subtelomeric loci found on chromosome VII was used to measure the enrichment of telomeric DNA in the immunoprecipitated, reverse-crosslinked and DNA-purified samples. The enrichment of telomeric sequence was expressed as TEL DNA immunoprecipitated relative to ARO1 and the fold change of the samples in each of the experiments are indicated as a relative quantity to control which is the value for either the 0 min time point (in synchronous experiments) or that of the untagged control (in asynchronous experiments). The results shown are an average of triplicates with error bars indicating standard deviations. The samples indicated in were all processed simultaneously.
TEL: Forward: 5′-TCATGTACGTCTCCTCCAAGCCC-3′
Reverse: 5′-GCAGTAGCGAGAGACAAGTGGGAAA-3′ARO1: Forward: 5′-TGACTGGTACTACCGTAACGGTTC-3′
Reverse: 5′- GAATACCATCTGGTAATTCTGTAGTTTTGAC-3′
Immunoprecipitation (IP)
The collection of cell cycle synchronous or asynchronous yeast cultures are as explained above except that the cultures are not crosslinked using formaldehyde. Immunoprecipitation assays were performed as described in.Citation28 It should be noted that phosphor-specific antibodies work best in IP protein lysates. The total Stn1 or Cdc13 is always indicated as the loading control in each Western blot shown in this study.
Fluorescence activated cell sorting (FACS)
Samples collected from yeast synchronization were fixed with 70% ethanol at 4°C overnight. The samples were then washed with 50mM sodium citrate (pH 7.5) and treated with 0.25mg/ml RNase A and 1.12mg/ml Proteinase K. The cells were then stained with SYBR® Green (Invitrogen) overnight, followed by sonication for 30 s on, 30 seconds off for 15 min using Diogenode Bioruptor® UCD-200. FACS was done using MACSQuant® VYB (Miltenyi Biotech) to obtain the DNA content of cell cycle synchronized cells, with a gate of 50,000 events. The analysis on the cellular DNA content obtained from the FACS was done using FlowJo VX.
Teloblot (southern blot using telomere-specific probe)
Yeast genomic DNA was prepared from yeast grown for about 100 generations using protocol as described in.Citation88 The genomic DNA was then digested with XhoI at 37°C for about 6 h following which the DNA was resolved using 0.8% agarose gel electrophoresis and transferred to Hybond-XL membrane (GE). The membrane was probed using a G-rich telomere-specific probe to analyze the C-rich telomeric sequence comprised of C(1–3)A. The probe used is a radiolabelled 32P-end-labeled oligonucleotide (5′-TGTGGTGTGTGGGTGTGGTGT-3′).
His-tag Purification for the kinase assay
pET28a-Stn1(M) – WT and the mutant with T223A,S250A, expressing 6xHis tagged recombinant Stn1(M) proteins, were used from.Citation28 For 6xHis tagged pET28a-Cdc13 and the phosphorylation mutants T308A or 7A, the plasmids were used from.Citation27
His-tagged Cdc13 and Stn1 recombinant proteins were expressed and batch purified from Tuner (DE3) Codon Plus bacteria (Novagen). The QiaExpressionist (Qiagen) protocol using Nickel-nitrilotriacetic acid (Ni-NTA) beads were used for the purification of the His-tagged proteins. The beads with the captured His-tag protein were resuspended in wash buffer which were resolved along with the flow through from the lysate and washes using SDS-PAGE, and the purified His-tagged protein was confirmed by Coomassie Blue (R-250) staining of the resolved gel and Western Blot Analysis using anti-His antibody.
TAP tag purification
TAP tag purification of the Cyclin-Cdk1-as1 complexes were performed in yeast strains harboring Δsic1, Cdk1-as1 and plasmid containing TAP-tagged cyclin under Gal-inducible promoter.Citation65,66 Overnight cultures grown in YEP (Yeast Extract-Peptone) and 2% Raffinose were diluted in large yeast cultures (10 L for Clb5, 4L for Clb3, 2L for Clb2) to OD600 of 0.1 and then grown to reach OD600 of 0.8 to 1.0, following which it was induced with galactose to a final concentration of 4% for about 4 h. The cultures were pelleted, lysed using TAP tag purification lysis buffer (25mM Hepes-HCL pH8.0, 1M NaCl, 0.1% NP-40, 1mM EDTA, 33mM EGTA, 1mM PMSF (1µg/Ml Aprotinin, Pepstatin A, Leupeptin), 50mM NaF, 80mM β-glycerophosphate, 1mM Na2VO3) in a bead beater with equal volume of glass beads added to the cell lysate suspension. The lysate was then cleared by ultra-centrifugation, spinning down at 50k rpm for 1 h at 4°C. The cleared lysate was then incubated with IgG beads for about 1 h at 4°C for the first affinity capture. Following washes with IPP150 buffer (25mM Hepes-HCL pH 8.0, 150 mM NaCl, 0.1% NP-40) the immunoprecipitated beads were incubated in TEV cleave buffer (25mM Hepes-HCL pH 8.0, 150 mM NaCl, 0.1% NP-40, 0.5mM EDTA, 1mM DTT). TEV protease was then added to the beads in the TEV cleavage buffer and mixed at room temperature for about 1–1.5 h. The cleaved proteins were then eluted and collected in a separate tube to which a final concentration of 1mM Mg Acetate, 1mM Imidazole and 2mM CaCl2 was added. This eluate was then incubated with calmodulin beads pre-washed in calmodulin binding buffer (10mM β-mercaptoethanol, 25mM Hepes-HCL pH 8.0, 150 mM NaCl, 1mM Mg Acetate, 1mM Imidazole and 2mM CaCl2) for about 1 h at 4°C. The protein-bound calmodulin beads were then washed with about 30ml calmodulin binding buffer before finally eluting the proteins using IPP150 Calmodulin Elution Buffer (10mM β-mercaptoethanol, 25mM Hepes-HCL pH 8.0, 150 mM NaCl, 1mM Mg Acetate, 1mM Imidazole and 2mM EGTA, 0.1% NP-40). Glycerol was added to the eluates at a final concentration of 15%.
In vitro kinase assay
For in vitro kinase assays, 5 to 10 µg of recombinant protein on Ni-NTA beads was incubated with normalized amounts of TAP-tag purified Cyclin-Cdk1-as1 complex, 1 mM creatine kinase-based ATP regeneration system, 1x kinase reaction buffer (25 mM Hepes, pH 7.4, 10 mM NaCl, and 2 mM MgCl2), 1x phosphatase inhibitors (50mMNaF, 80mM β-glycerophosphate, and 1mMNa3VO4), and 5 to 10 µCi N6-(benzyl)-[γ-32P]ATP in a final 30µl volume at room temperature for 2 time points - 30 min and 1 h. The normalizing amounts of Clb5/3/2-Cdk1 complex incubated with Cdc13 or Stn1 were based on the in vitro phosphorylation values of Histone (Figure S4) that is consistent with Koivomagi et al., 2011 [53]. The His-tagged recombinant proteins were resolved by SDS-PAGE, stained with Coomassie brilliant blue R-250, and exposed in a PhosphoImager cassette overnight. While the protein input levels were obtained from the coomassie stained image, the phosphorylation levels were obtained from the autoradiograph obtained from scanning the PhosphoImage using ImageQuant (Molecular Dynamics). The intensity of the corresponding bands were quantified using ImageJ software. The relative phosphorylation efficiency of WT over mutants were quantified and normalized relative to input protein levels.
Intensity (WT/Mut) = [(Phosphorylation Intensity/Input)WT]/[(Phosphorylation Intensity/Input)mut]
Disclosure of potential conflicts of interest
The authors have no potential conflict of interest to be reported.
Author contributions
V. G. and S. L. conceived and coordinates the study and wrote the paper. V.G. and C.R.T. performed and analyzed the experiments. All authors reviewed the data and approved the final version of the manuscript.
Supplemental_Material.pdf
Download PDF (7.2 MB)Acknowledgments
We thank Dr. Uttam Surana and Dr. Hong Hwa Lim for technical support, invaluable discussions and critical reading of this manuscript. We thank Dr. Mardo Koivomagi for his helpful suggestions on purification of Cyclin-Cdk1 complexes and in vitro kinase assays.
Funding
The study is funded by MOE Tier 2 grant to S.L.
References
- Muller, H. The remaking of chromosomes. Collecting net 1938; 13(198):181-95.
- McClintock, B. The Fusion of Broken Ends of Chromosomes Following Nuclear Fusion. Proc Natl Acad Sci U S A 1942; 28(11):458-63; PMID:16578057; https://doi.org/10.1073/pnas.28.11.458
- Blackburn, EH. Structure and function of telomeres. Nature 1991; 350(6319):569-73; PMID:1708110; https://doi.org/10.1038/350569a0
- Carneiro, T, Khair, L, Reis, CC, Borges, V, Moser, BA, Nakamura, TM, Ferreira, MG. Telomeres avoid end detection by severing the checkpoint signal transduction pathway. Nature 2010; 467(7312):228-32; https://doi.org/10.1038/nature09353
- Blackburn, EH, Gall, JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol 1978; 120(1):33-53; PMID:642006; https://doi.org/10.1016/0022-2836(78)90294-2
- Blackburn, EH, Greider, CW, Szostak, JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med 2006; 12(10):1133-8; PMID:17024208; https://doi.org/10.1038/nm1006-1133
- Shampay, J, Szostak, JW, Blackburn, EH. DNA sequences of telomeres maintained in yeast. Nature 1984; 310(5973):154-7; PMID:6330571; https://doi.org/10.1038/310154a0
- Linger, BR, Price, CM. Conservation of telomere protein complexes: shuffling through evolution. Crit Rev Biochem Mol Biol 2009; 44(6):434-46; PMID:19839711; https://doi.org/10.3109/10409230903307329
- Watson, JD. Origin of concatemeric T7DNA. Nature 1972; 239(94):197-201; PMID:4263504
- Verdun, RE, Karlseder, J. Replication and protection of telomeres. Nature 2007; 447(7147):924-31; PMID:17581575; https://doi.org/10.1038/nature05976
- Gilson, E, Geli, V. How telomeres are replicated. Nat Rev Mol Cell Biol 2007; 8(10):825-38; PMID:17885666; https://doi.org/10.1038/nrm2259
- Greider, CW, Blackburn, EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985; 43(2 Pt 1):405-13; PMID:3907856; https://doi.org/10.1016/0092-8674(85)90170-9
- Greider, CW, Blackburn, EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 1987; 51(6):887-98; PMID:3319189; https://doi.org/10.1016/0092-8674(87)90576-9
- Fan, X, Price, CM. Coordinate regulation of G-and C strand length during new telomere synthesis. Mol Biol Cell 1997; 8(11):2145-55; PMID:9362059; https://doi.org/10.1091/mbc.8.11.2145
- Armanios, M, Blackburn, EH. The telomere syndromes. Nat Rev Genet 2012; 13(10):693-704; PMID:22965356; https://doi.org/10.1038/nrg3246
- Holohan, B, Wright, WE, Shay, JW. Telomeropathies: An emerging spectrum disorder. J Cell Biol 2014; 205(3):289-99; PMID:24821837; https://doi.org/10.1083/jcb.201401012
- Borah, S, Xi, L, Zaug, AJ, Powell, NM, Dancik, GM, Cohen, SB, Costello, JC, Theodorescu, D, Cech, TR. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015; 347(6225):1006-10; https://doi.org/10.1126/science.1260200
- Shay, J, Bacchetti, S. A survey of telomerase activity in human cancer. Eur J Cancer 1997; 33(5):787-91; PMID:9282118; https://doi.org/10.1016/S0959-8049(97)00062-2
- Hanahan, D, Weinberg, RA. Hallmarks of cancer: the next generation. Cell 2011; 144(5):646-74; PMID:21376230; https://doi.org/10.1016/j.cell.2011.02.013
- Polvi, A, Linnankivi, T, Kivelä, T, Herva, R, Keating, JP, Mäkitie, O, Pareyson, D, Vainionpää, L, Lahtinen, J, Hovatta, I, et al. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum Genet 2012; 90(3):540-9; https://doi.org/10.1016/j.ajhg.2012.02.002
- Anderson, BH, Kasher, PR, Mayer, J, Szynkiewicz, M, Jenkinson, EM, Bhaskar, SS, Urquhart, JE, Daly, SB, Dickerson, JE, O'Sullivan, J, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet 2012; 44(3):338-42; https://doi.org/10.1038/ng.1084
- Chen, LY, Majerska, J, Lingner, J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev 2013; 27(19):2099-108; PMID:24115768; https://doi.org/10.1101/gad.222893.113
- Keller, RB, Gagne, KE, Usmani, GN, Asdourian, GK, Williams, DA, Hofmann, I, Agarwal, S. CTC1 Mutations in a patient with dyskeratosis congenita. Pediatric Blood Cancer 2012; 59(2):311-4; https://doi.org/10.1002/pbc.24193
- Gu, P, Min, JN, Wang, Y, Huang, C, Peng, T, Chai, W, Chang, S. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J 2012; 31(10):2309-21; https://doi.org/10.1038/emboj.2012.96
- Walne, AJ, Bhagat, T, Kirwan, M, Gitiaux, C, Desguerre, I, Leonard, N, Nogales, E, Vulliamy, T, Dokal, IS. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica 2013; 98(3):334-8; PMID:22899577; https://doi.org/10.3324/haematol.2012.071068
- Frank, CJ, Hyde, M, Greider, CW. Regulation of telomere elongation by the cyclin-dependent kinase CDK1. Mol Cell 2006; 24(3):423-32; PMID:17070718; https://doi.org/10.1016/j.molcel.2006.10.020
- Li, S, Makovets, S, Matsuguchi, T, Blethrow, JD, Shokat, KM, Blackburn, EH. Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell 2009; 136(1):50-61; https://doi.org/10.1016/j.cell.2008.11.027
- Liu, C-C, Gopalakrishnan, V, Poon, LF, Yan, T, Li, S. Cdk1 regulates the temporal recruitment of telomerase and Cdc13-Stn1-Ten1 complex for telomere replication. Mol Cell Biol 2014; 34(1):57-70; PMID:24164896; https://doi.org/10.1128/MCB.01235-13
- Diede, SJ, Gottschling, DE. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases α and δ. Cell 1999; 99(7):723-33; PMID:10619426; https://doi.org/10.1016/S0092-8674(00)81670-0
- Zhu, Z, Chung, WH, Shim, EY, Lee, SE, Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008; 134(6):981-94; https://doi.org/10.1016/j.cell.2008.08.037
- Hughes, TR, Evans, SK, Weilbaecher, RG, Lundblad, V. The Est3 protein is a subunit of yeast telomerase. Current Biology 2000; 10(13):809-12; https://doi.org/10.1016/S0960-9822(00)00562-5
- Zhou, J, Hidaka, K, Futcher, B. The Est1 subunit of yeast telomerase binds the Tlc1 telomerase RNA. Mol Cell Biol 2000; 20(6):1947-55; PMID:10688642; https://doi.org/10.1128/MCB.20.6.1947-1955.2000
- Counter, CM, Meyerson, M, Eaton, EN, Weinberg, RA. The catalytic subunit of yeast telomerase. Proc Natl Acad Sci U S A 1997; 94(17):9202-7; https://doi.org/10.1073/pnas.94.17.9202
- Nugent, CI, Hughes, TR, Lue, NF, Lundblad, V. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science 1996; 274(5285):249-52; https://doi.org/10.1126/science.274.5285.249
- Grandin, N, Reed, SI, Charbonneau, M. Stn1, a new Saccharomyces cerevisiae protein, is implicated in telomere size regulation in association with Cdc13. Genes Dev 1997; 11(4):512-27; PMID:9042864; https://doi.org/10.1101/gad.11.4.512
- Grandin, N, Damon, C, Charbonneau, M. Ten1 functions in telomere end protection and length regulation in association with Stn1 and Cdc13. EMBO J 2001; 20(5):1173-83; PMID:11230140; https://doi.org/10.1093/emboj/20.5.1173
- Gao, H, Cervantes, RB, Mandell, EK, Otero, JH, Lundblad, V. RPA-like proteins mediate yeast telomere function. Nat Struct Mol Biol 2007; 14(3):208-14; https://doi.org/10.1038/nsmb1205
- Qi, H, Zakian, VA. The Saccharomyces telomere-binding protein Cdc13p interacts with both the catalytic subunit of DNA polymerase alpha and the telomerase-associated est1 protein. Genes Dev 2000; 14(14):1777-88; PMID:10898792
- Chandra, A, Hughes, TR, Nugent, CI, Lundblad, V. Cdc13 both positively and negatively regulates telomere replication. Genes Dev 2001; 15(4):404-14; https://doi.org/10.1101/gad.861001
- Petreaca, RC, Chiu, HC, Eckelhoefer, HA, Chuang, C, Xu, L, Nugent, CI. Chromosome end protection plasticity revealed by Stn1p and Ten1p bypass of Cdc13p. Nat Cell Biol 2006; 8(7):748-55; PMID:16767082; https://doi.org/10.1038/ncb1430
- Weinert, T, Hartwell, LH. The RAD9 gene controls the cell cycle response to DNA damage in. Saccharomyces cerevisiae Science 1988; 241:317-22; PMID:3291120
- Garvik, B, Carson, M, Hartwell, L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol 1995; 15(11):6128-38; PMID:7565765; https://doi.org/10.1128/MCB.15.11.6128
- Martin, V, Du, LL, Rozenzhak, S, Russell, P. Protection of telomeres by a conserved Stn1-Ten1 complex. Proc Natl Acad Sci U S A 2007; 104(35):14038-43; https://doi.org/10.1073/pnas.0705497104
- Puglisi, A, Bianchi, A, Lemmens, L, Damay, P, Shore, D. Distinct roles for yeast Stn1 in telomere capping and telomerase inhibition. EMBO J 2008; 27(17):2328-39; PMID:19172739; https://doi.org/10.1038/emboj.2008.158
- Grandin, N, Damon, C, Charbonneau, M. Cdc13 prevents telomere uncapping and Rad50-dependent homologous recombination. EMBO J 2001; 20(21):6127-39; PMID:11689452; https://doi.org/10.1093/emboj/20.21.6127
- Pennock, E, Buckley, K, Lundblad, V. Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell 2001; 104(3):387-96; PMID:11239396; https://doi.org/10.1016/S0092-8674(01)00226-4
- Vodenicharov, MD, Wellinger, RJ. DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol Cell 2006; 24(1):127-37; PMID:17018298; https://doi.org/10.1016/j.molcel.2006.07.035
- Bonetti, D, Martina, M, Clerici, M, Lucchini, G, Longhese, MP. Multiple pathways regulate 3′ overhang generation at S. Cerevisiae Telomeres. Mol Cell 2009; 35(1):70-81; https://doi.org/10.1016/j.molcel.2009.05.015
- Zubko, MK, Guillard, S, Lydall, D. Exo1 and Rad24 differentially regulate generation of ssDNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics 2004; 168(1):103-15; PMID:15454530; https://doi.org/10.1534/genetics.104.027904
- Loog, M, Morgan, DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature 2005; 434(7029):104-8; PMID:15744308; https://doi.org/10.1038/nature03329
- Miller, ME, Cross, FR. Cyclin specificity: how many wheels do you need on a unicycle?. J Cell Sci 2001; 114(10):1811-20; PMID:11329367
- Roberts, JM. Evolving ideas about cyclins. Cell 1999; 98(2):129-32; PMID:10428024; https://doi.org/10.1016/S0092-8674(00)81007-7
- Koivomagi, M, Valk, E, Venta, R, Iofik, A, Lepiku, M, Morgan, DO, Loog, M. Dynamics of Cdk1 substrate specificity during the cell cycle. Mol Cell 2011; 42(5):610-23; PMID:21658602; https://doi.org/10.1016/j.molcel.2011.05.016
- Kitagawa, M, Higashi, H, Jung, HK, Suzuki-Takahashi, I, Ikeda, M, Tamai, K, Kato, J, Segawa, K, Yoshida, E, Nishimura, S, et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J 1996; 15(24):7060
- Brown, NR, Noble, ME, Endicott, JA, Johnson, LN. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat Cell Biol 1999; 1(7):438-43; https://doi.org/10.1038/15674
- Coudreuse, D, Nurse, P. Driving the cell cycle with a minimal CDK control network. Nature 2010; 468(7327):1074-79; PMID:21179163; https://doi.org/10.1038/nature09543
- Stern, B, Nurse, P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends in Genetics 1996; 12(9):345-50; PMID:8855663; https://doi.org/10.1016/S0168-9525(96)80016-3
- Fisher, D, Krasinska, L, Coudreuse, D, Novák, B. Phosphorylation network dynamics in the control of cell cycle transitions. J Cell Sci 2012; 125(20):4703-11; https://doi.org/10.1242/jcs.106351
- Fisher, D, Nurse, P. A single fission yeast mitotic cyclin B p34cdc2 kinase promotes both S-phase and mitosis in the absence of G1 cyclins. EMBO J 1996; 15(4):850; PMID:8631306
- Schwob, E, Nasmyth, K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev 1993; 7(7a):1160-75; https://doi.org/10.1101/gad.7.7a.1160
- Fitch, I, Dahmann, C, Surana, U, Amon, A, Nasmyth, K, Goetsch, L, Byers, B, Futcher, B. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol Biol Cell 1992; 3(7):805-18; https://doi.org/10.1091/mbc.3.7.805
- Surana, U, Robitsch, H, Price, C, Schuster, T, Fitch, I, Futcher, AB, Nasmyth, K. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell 1991; 65(1):145-61; PMID:1849457; https://doi.org/10.1016/0092-8674(91)90416-V
- Marcand, S, Brevet, V, Mann, C, Gilson, E. Cell cycle restriction of telomere elongation. Current Biology 2000; 10(8):487-490; PMID:10801419; https://doi.org/10.1016/S0960-9822(00)00450-4
- Elledge, SJ. Cell cycle checkpoints: preventing an identity crisis. Science 1996; 274(5293):1664; PMID:8939848; https://doi.org/10.1126/science.274.5293.1664
- Puig, O, Caspary, F, Rigaut, G, Rutz, B, Bouveret, E, Bragado-Nilsson, E, Wilm, M, Séraphin, B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 2001; 24(3):218-29; PMID:11403571; https://doi.org/10.1006/meth.2001.1183
- Ubersax, JA, Woodbury, EL, Quang, PN, Paraz, M, Blethrow, JD, Shah, K, Shokat, KM, Morgan, DO. et al. Targets of the cyclin-dependent kinase Cdk1. Nature 2003; 425(6960):859-64; PMID:14574415; https://doi.org/10.1038/nature02062
- Kõivomägi, M, Ord, M, Iofik, A, Valk, E, Venta, R, Faustova, I, Kivi, R, Balog, ER, Rubin, SM, Loog, M. Multisite phosphorylation networks as signal processors for Cdk1. Nat Struct Mol Biol 2013; 20(12):1415-1424; https://doi.org/10.1038/nsmb.2706
- Visintin, R, Craig, K, Hwang, ES, Prinz, S, Tyers, M, Amon, A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell 1998; 2(6):709-18; PMID:9885559; https://doi.org/10.1016/S1097-2765(00)80286-5
- Surana, U, Amon, A, Dowzer, C, McGrew, J, Byers, B, Nasmyth, K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J 1993; 12(5):1969 PMID:8491189
- Epstein, CB, Cross, FR. CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev 1992; 6(9):1695-706; https://doi.org/10.1101/gad.6.9.1695
- Donaldson, AD. The yeast mitotic cyclin Clb2 cannot substitute for S phase cyclins in replication origin firing. EMBO Rep 2000 l; 1(6):507-12; PMID:11263495; https://doi.org/10.1093/embo-reports/kvd108
- Lew, DJ, Reed, SI. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J Cell Biol 1995; 129(3):739-49; PMID:7730408; https://doi.org/10.1083/jcb.129.3.739
- Donaldson, AD, Raghuraman, MK, Friedman, KL, Cross, FR, Brewer, BJ, Fangman, WL. CLB5-dependent activation of late replication origins in S. Cerevisiae. Mol Cell 1998; 2(2):173-82; https://doi.org/10.1016/S1097-2765(00)80127-6
- Shen, ZJ, Hsu, PH, Su, YT, Yang, CW, Kao, L, Tseng, SF, Tsai, MD, Teng, SC. PP2A and Aurora differentially modify Cdc13 to promote telomerase release from telomeres at G2/M phase. Nat Commun 2014; 5:5312; https://doi.org/10.1038/ncomms6312
- Kupiec, M. Biology of telomeres: lessons from budding yeast. FEMS Microbiol Rev 2014; 38(2):144-71; PMID:24754043; https://doi.org/10.1111/1574-6976.12054
- Jin, F, Liu, H, Liang, F, Rizkallah, R, Hurt, MM, Wang, Y. Temporal control of the dephosphorylation of Cdk substrates by mitotic exit pathways in budding yeast. Proc Nat Acad Sci 2008; 105(42):16177-82; https://doi.org/10.1073/pnas.0808719105
- Grossi, S, Puglisi, A, Dmitriev, PV, Lopes, M, Shore, D. Pol12, the B subunit of DNA polymerase α, functions in both telomere capping and length regulation. Genes Dev 2004; 18(9):992-1006; https://doi.org/10.1101/gad.300004
- Grossi, S, Puglisi, A, Dmitriev, PV, Lopes, M, Shore, D. Pol12, the B subunit of DNA polymerase alpha, functions in both telomere capping and length regulation. Genes Dev 2004; 18(9):992-1006; PMID:15132993; https://doi.org/10.1101/gad.300004
- Bouchoux, C, Uhlmann, F. A quantitative model for ordered Cdk substrate dephosphorylation during mitotic exit. Cell 2011; 147(4):803-14; PMID:22078879; https://doi.org/10.1016/j.cell.2011.09.047
- Uhlmann, F, Bouchoux, C, López-Avilés, S. A quantitative model for cyclin-dependent kinase control of the cell cycle: revisited. Phil. Trans. R. Soc. B 2011; 366(1584):3572-3583; https://doi.org/10.1098/rstb.2011.0082
- Porter, AC. Preventing DNA over-replication: a Cdk perspective. Cell Division 2008; 3(1):1-10; PMID:18179693; https://doi.org/10.1186/1747-1028-3-3
- Queralt, E, Uhlmann, F. Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol 2008; 20(6):661-8; PMID:18845253; https://doi.org/10.1016/j.ceb.2008.09.003
- Satyanarayana, A, Kaldis, P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009; 28(33):2925-39; PMID:19561645; https://doi.org/10.1038/onc.2009.170
- Turner, NC, Ro, J, André, F, Loi, S, Verma, S, Iwata, H, Harbeck, N, Loibl, S, Huang Bartlett, C, Zhang, K, et al. Palbociclib in hormone-receptor–positive advanced breast cancer. N Engl J Med 2015; 373(3):209-19; https://doi.org/10.1056/NEJMoa1505270
- Finn, RS, Crown, JP, Lang, I, Boer, K, Bondarenko, IM, Kulyk, SO, Ettl, J, Patel, R, Pinter, T, Schmidt, M, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015; 16(1):25-35; PMID:25524798; https://doi.org/10.1016/S1470-2045(14)71159-3
- Longtine, MS, McKenzie, A 3rd, Demarini, DJ, Shah, NG, Wach, A, Brachat, A, Philippsen, P, Pringle, JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998; 14(10):953-61; PMID:9717241; https://doi.org/10.1002/(SICI)1097-0061(199807)14:10%3c953::AID-YEA293%3e3.0.CO;2-U
- Janke, C, Magiera, MM, Rathfelder, N, Taxis, C, Reber, S, Maekawa, H, Moreno-Borchart, A, Doenges, G, Schwob, E, Schiebel, E, et al. A versatile toolbox for PCR‐based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 2004; 21(11):947-62; PMID:15334558; https://doi.org/10.1002/yea.1142
- Lundblad, V, Szostak, JW. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 1989; 57(4):633-43; PMID:2655926; https://doi.org/10.1016/0092-8674(89)90132-3