
Viruses are known to interfere with the DNA damage response (DDR) of host cells. Adenoviruses, for example target elements of non-homologous end joining (NHEJ) for ubiquitination to prevent concatemerization of the linear viral genomes.Citation1 At the same time, DDR factors are recruited to adenovirus replication centers to assist in viral DNA replication.Citation2 Viruses in the Retroviridae family also target DDR elements. While HIV-1, through its accessory protein, Vpr, induces a DNA damage signal that has the hallmarks of replication stress, the Tax protein from HTLV-I attenuates DNA damage signals to suppress cell cycle checkpoint activation (reviewed inCitation3).
A unique feature of retroviruses is their ability to form a DNA copy of their RNA genome, which is then permanently inserted in the host chromosome to form a provirus. Following integration, retroviruses continue their life cycle by initiating viral transcription. Occasionally, a retrovirus may fail to elicit viral gene expression, leading to a state that can be regarded as latency. Latency is thought to be major hurdle toward HIV-1 eradication in patients, and failure to transcribe can result from a variety of mechanisms, including entry of cells in a quiescent state, viral integration into transcription-poor regions, low abundance of transcription factors and transcriptional interference with neighboring genes, among others. Because of the lack of viral gene expression, T cells harboring latent viruses are indistinguishable from their uninfected counterparts. Thus, any properties that may distinguish latently infected cells from uninfected ones are hotly sought after. Piekna-Przybylska and collaborators in an article in this volume of Cell CycleCitation4 set out to test whether HIV-1 latently infected cells are different from uninfected cells in their response to genotoxic stress.
The authors exposed cells to several bona fide genotoxic agents such as high concentrations of nucleoside-analog reverse transcriptase inhibitors (NRTIs) and etoposide (an inhibitor of topoisomerase II), and directly assessed DNA damage by detection of the phosphorylated forms of histone H2A-X (γ-H2A-X; Ser139), ATM(Ser1981), Chk2(Thr68) and p53(Ser15). Uninfected Jurkat cells displayed an inherent propensity to DNA damage typical of transformed cells, as evidenced by the high percentage of γ-H2A-X at baseline, which was exacerbated when cells were exposed to etoposide. EF7 and CA5 cells, Jurkat derivatives harboring single copies of latent HIV-1,Citation5 displayed a 2-to-6-fold increased level of γH2A-X, indicating a propensity to spontaneous DNA damage in the form of double-stranded breaks (DSB). Accordingly, when EF7 and CA5 cells were challenged with etoposide, virtually all cells in culture turned positive for γH2A-X after 2 hours of exposure. Furthermore, individual cells displayed almost one order of magnitude-increased higher mean fluorescence intensity for γH2A-X, indicating a higher frequency of DSB per cell in the case of latent infection.
To validate their observations in primary cells, devoid of the inherent genomic instability that characterizes transformed cells, the authors generated latently infected central memory T cells in vitro, and challenged them with etoposide, an inhibitor of topoisomerase II. Here, etoposide by itself induced significant DNA damage as evidenced by the presence of γH2A-X in 15–18% of uninfected primary cells. In latently infected cells from 7 different donors, the same treatment generated several fold higher levels of γH2A-X.
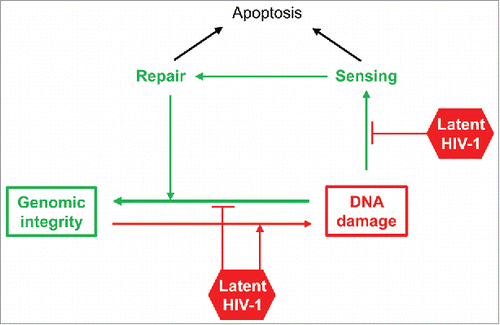
The above study therefore puts forward the tantalizing possibility that the presence of HIV-1, even in the absence of detectable viral gene expression, may be sufficient to increase the host cell propensity to undergo DNA damage. Since DNA damage, its sensing, and its repair are interdependent (see ), latent infection of T cells may potentially have effects on all 3 processes. Thus, the presence of a latent virus could directly promote increased frequency of DSB, but also defective sensing of DNA damage as well as defective repair mechanisms. The final result would seemingly be same: increased accumulation of DNA damage.
The observations by Piekna-Przybylska are certainly provocative for several reasons. First, one must ponder how a “silent” virus can affect the genomic stability of the host cell. Several potential explanations may be appropriate here. Latent HIV may not be totally silent, and perhaps expression of low levels of viral RNAs may represent a genomic threat to cells. It is also possible that prior expression of a viral genotoxic protein (such as Vpr) may have set a DDR in motion, which persists after the viral protein is no longer present. In partial support of this idea, a recent publication reports that latently infected cells have a transcriptomic profile that is enriched for expression of genes related to p53 signaling.Citation6
A second question relates to the potential consequences for therapy: can we exploit the enhanced propensity to DNA damage as a means for selectively inducing death in these cells? The observations by Piekna-Przybylska suggest that in vitro this may be the case and therefore set up the stage for future explorations.
Figure 1. Potential mechanisms by which latent HIV-1 could contribute to DNA damage. Activities leading to genotoxic stress are depicted in red; activities leading to genomic stability are in green.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Weiden MD, Ginsberg HS. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc Natl Acad Sci U S A 1994; 91:153-7; PMID:8278357; https://doi.org/10.1073/pnas.91.1.153
- Stracker TH, Lee DV, Carson CT, Araujo FD, Ornelles DA, Weitzman MD. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J Virol 2005; 79:6664-73; PMID:15890904; https://doi.org/10.1128/JVI.79.11.6664-6673.2005
- Ryan EL, Hollingworth R, Grand RJ. Activation of the DNA Damage Response by RNA Viruses. Biomolecules 2016; 6:2; PMID:26751489; https://doi.org/10.3390/biom6010002
- Piekna-Przybylska D, Sharma G, Maggirwar SB, Bambara RA. Deficiency in DNA damage response, a new characteristic of cells infected with latent HIV-1. Cell Cycle 2017; 16(10):968-978; PMID:28388353; https://doi.org/10.1080/15384101.2017.1312225
- Duverger A, Jones J, May J, Bibollet-Ruche F, Wagner FA, Cron RQ, Kutsch O. Determinants of the establishment of human immunodeficiency virus type 1 latency. J Virol 2009; 83:3078-93; PMID:19144703; https://doi.org/10.1128/JVI.02058-08;JVI.02058-08[pii]
- White CH, Moesker B, Beliakova-Bethell N, Martins LJ, Spina CA, Margolis DM, Richman DD, Planelles V, Bosque A, Woelk CH. Transcriptomic Analysis Implicates the p53 Signaling Pathway in the Establishment of HIV-1 Latency in Central Memory CD4 T Cells in an In Vitro Model. PLoS Pathog 2016; 12:e1006026; PMID:27898737; https://doi.org/10.1371/journal.ppat.1006026