
ABSTRACT
The centromere plays an essential role in accurate chromosome segregation, and defects in its function lead to aneuploidy and thus cancer. The centromere-specific histone H3 variant CENP-A is proposed to be the epigenetic mark of the centromere, as active centromeres require CENP-A–containing nucleosomes to direct the recruitment of multiple kinetochore proteins. CENP-A K124 ubiquitylation, mediated by CUL4A-RBX1-COPS8 E3 ligase activity, is required for CENP-A deposition at the centromere. However, the mechanism that controls the E3 ligase activity of the CUL4A-RBX1-COPS8 complex remains obscure. We have discovered that the SGT1-HSP90 complex is required for recognition of CENP-A by COPS8. Thus, the SGT1-HSP90 complex contributes to the E3 ligase activity of the CUL4A complex that is necessary for CENP-A ubiquitylation and CENP-A deposition at the centromere.
Introduction
During vertebrate cell division, the genome must be segregated equally between the daughter cells, and the mitotic spindle must attach to a single locus on each chromosome, termed the centromere, to ensure the stable transmission of the genome. In most eukaryotes, centromeres are specified epigenetically by the presence of the histone H3 variant CENP-A. Centromere inheritance depends on the maintenance of CENP-A-containing nucleosomes at a single site on each chromosome.Citation1,2 CENP-A–containing nucleosomes localize to the inner plate of mammalian kinetochoresCitation3 and bind to the 171-bp α-satellite DNA in humans.Citation4 Active centromeres require CENP-A–containing nucleosomes to direct the recruitment of a constitutive centromere-associated network (CCAN) and the kinetochore proteins, which together orchestrate the kinetochore-microtubule attachment and regulate cycle progression through the spindle checkpoint.Citation5 Centromere inheritance is presumably determined by the spatial and temporal architecture of the centromeric nucleosome, but the mechanism by which inheritance occurs is largely unknown. We previously reported that CENP-A K124 ubiquitylation, which is mediated by CUL4A-RBX1-COPS8 E3 ligase activity, is required for CENP-A deposition at the centromere.Citation6
The kinetochore consists of centromeric DNA elements and structural proteins. Using a variety of approaches, recent studies of vertebrate cells have identified and characterized multiple kinetochore proteins assembled on centromeric DNA to form the kinetochore structure.Citation5 CCAN proteins create a platform for kinetochore formation and subsequently recruit outer kinetochore components such as KNL1, the MIS12 complex, and the NDC80 complex (the KMN network).Citation7 KMN network components that attach to the spindle microtubules comprise the functional kinetochore together.Citation8
Yeast SGT1 (the homolog of SGT1/SUGT1 in humans) was originally identified not only as a dosage suppressor of the skp1–4 mutation, which causes defects in yeast kinetochore function, but also as a novel subunit of the Skp1-Cullin-F-box (SCF) ubiquitin ligase complex.Citation9 Budding yeast Sgt1 binds to Skp1 to assemble the CBF3 complex (the core component of the kinetochore) via Ctf13 activation,Citation9,10 and Hsp90 is also required for the CBF3 assembly.Citation11 HSP90 is a molecular chaperone involved in the folding, assembly, maturation, and stabilization of specific target proteins (often called “HSP90 clients”), and HSP90 performs these functions in different complexes containing various cochaperones.Citation12-14 Cochaperones, which interact with and are required for HSP90 function, regulate the ATPase activity of HSP90 and recruit client proteins to HSP90.Citation15 As these functions are derived from the binding of SGT1 to HSP90, SGT1/SUGT1 is a co-chaperone.Citation16-20 SGT1 is involved in multiple specific cellular functions including ubiquitylation, cyclic AMP pathway, centrosome maturation, kinetochore assembly, immune response, neuroblast cortical polarity, epithelial morphogenesis, and oncogenesis, presumably as a cochaperone.Citation21 In humans, the SGT1-HSP90 complex is required for kinetochore assemblyCitation22-24 and participates in kinetochore-microtubule attachment by stabilizing the MIS12 complex at kinetochores.Citation22,25 However, the mechanism to recruit multiple kinetochore and spindle checkpoint proteins, in particular, the mechanism by which the SGT1-HSP90 complex recruites inner kinetochore proteins in humans, has not yet been clearly demonstrated.
We report here that the results of RNA interference (RNAi)-mediated SGT1 and/or HSP90 depletion in HeLa cells revealed an essential role of the SGT1-HSP90 complex in kinetochore assembly. The SGT1-HSP90 complex is required for CENP-A ubiquitylation in vivo and CENP-A deposition at centromeres. We here clarify how the SGT1-HSP90 complex contributes to the E3 ligase activity of the CUL4A complex in CENP-A ubiquitylation. Our results demonstrate that the SGT1-HSP90 complex is required for recognition of CENP-A by COPS8.
Results
SGT1 is Required for Localization of CENP-A to Centromeres
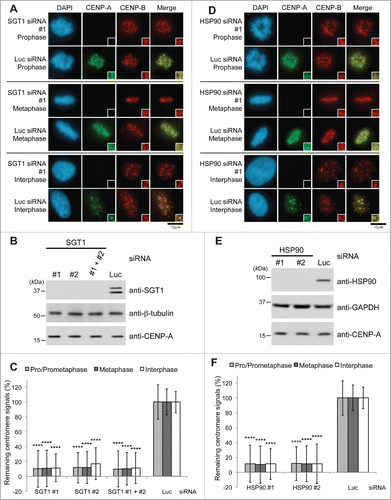
Previous studies implicated SGT1 as an essential protein and a critical factor for mammalian kinetochore assemblyCitation24; therefore, we hypothesized that SGT1 plays a role in the localization of CENP-A to centromeres. To determine whether SGT1 functions in CENP-A loading on centromeres, we first knocked down expression of SGT1 by using siRNA, and this approach led to the silencing of both SGT1 isoforms (A and B isoforms) in HeLa cells.Citation26 Depletion of SGT1 significantly induced a reduction of CENP-A immunofluorescence signals at centromeres 72 h after transfection (; Fig. S1A). SGT1 levels in total cell lysates were greatly reduced, but CENP-A levels remained unchanged at the same time point (). By confirming that the protein levels of CENP-A in total cell lysates were similar to those in lysates of cells transfected with luciferase (Luc) siRNA under the same culture conditions (), we ruled out the possibility that SGT1 depletion causes CENP-A protein degradation. The number of abnormal metaphase cells was significantly increased by SGT1 depletion (Fig. S6A and B). These results are consistent with those of a previous study that showed that depletion of SGT1 reduces the centromeric localization of inner-central and outer kinetochore proteins (CENP-H, CENP-I, CENP-K, CENP-N, CENP-U, the MIS12 complex, and the NDC complex)Citation22,24; these earlier results indicated that CENP-A is at the top of a hierarchy of the pathway that determines the assembly of kinetochore components and is required for recruitment of these kinetochore proteins to the kinetochore.Citation27
Figure 1. SGT1-HSP90 complex is required for CENP-A localization at centromeres. (A) SGT1 siRNA causes delocalization of CENP-A from centromeres. HeLa cells were transfected for 72 h with SGT1 siRNAs (targeting both A and B isoforms) or luciferase (Luc) siRNA control (Table S2). Immunostaining with DAPI (blue), CENP-A (green), and CENP-B (red) during prophase, metaphase, and interphase. Scale bar, 10 μm. (B) Western blot analysis of total HeLa cell lysates harvested 72 h after transfection with SGT1 siRNA (targeting both A and B isoforms) or Luc siRNA control (Table S2) revealed depletion of SGT1, but level of CENP-A protein remained unchanged. β-tubulin protein was the loading control. (C) CENP-A signals at centromeres shown in (A) were quantified. Signals were normalized to those of Luc siRNA-treated cells, and the mean percentages ( ± SD) are shown. (****) P < 0.0001 compared with signals of Luc siRNA-treated cells in each immunostaining experiment (Student's t test). (D) HSP90 siRNA causes delocalization of CENP-A from centromeres. HeLa cells were transfected for 72 h with HSP90 siRNAs (targeting both α and β isoforms) or Luc siRNA control (Table S2). Immunostaining with DAPI (blue), CENP-A (green), and CENP-B (red) during prophase, metaphase, and interphase. Scale bar, 10 μm. (E) Western blot analysis of total HeLa cell lysates harvested 72 h after transfection with HSP90 siRNA (targeting both α and β isoforms) or Luc siRNA control (Table S2) revealed depletion of HSP90, but the level of CENP-A protein remained unchanged. GAPDH protein was the loading control. (F) CENP-A signals at centromeres shown in (D) were quantified. Signals were normalized to those of Luc siRNA-treated cells, and the mean percentages ( ± SD) are shown. (****) P < 0.0001 compared with Luc siRNA-treated cells (Student's t test).
SGT1-HSP90 complex is required for localization of CENP-A to centromeres
SGT1 interacts with HSP90 in human cells, and depletion using HSP90α− and HSP90β−specific siRNA together but not individually prevents the centromeric localization of CENP-H, CENP-I, and BUB1 but not of CENP- B and CENP-C.Citation23 Therefore, we hypothesized that HSP90 also plays a role in the localization of CENP-A to centromeres. In the present study, siRNA-mediated depletion of HSP90 led to a reduction in CENP-A immunofluorescence signals at centromeres (; Fig. S1B; #1 and #2 targeted both HSP90α and HSP90β together; Table S2). CENP-A levels in total cell lysates remained unchanged at the same time point (), and this absence of change ruled out the possibility of CENP-A protein degradation that resulted from HSP90 depletion. Depletion of HSP90 did not induce a reduction of protein level of central kinetochore components (CENP-H and CENP-I), but immunofluorescence signals of these components at centromeres were reduced concomitantly as shown in our previous studyCitation23 (data not shown). The number of abnormal metaphase cells was significantly increased by HSP90 depletion (Fig. S6A and B).
We previously found that the HSP90 inhibitor 17-AAG causes delocalization of central and outer kinetochore proteins (CENP-H, CENP-I, CENP-E, and HEC1) and spindle checkpoint components (BUB1 and BUBR1), although the centromeric localization of CENP-B and CENP-C was not substantially affected.Citation23 In the present study, we used the same range of 17-AAG concentrations (10, 100, 500, 800, and 1000 nM) for 24 h and found that CENP-A delocalized from centromeres (Fig. S2A–C). The amounts of CENP-A protein in cell lysates were not substantially reduced (Fig. S2B), but CENP-A signals at centromeres were substantially reduced in a dose-dependent manner in 17-AAG–treated HeLa cells (Fig. S2C). Because 17-AAG binds to the ATP-binding pocket of HSP90, inhibits its ATPase activity, and inhibits chaperone function for HSP90 client proteinsCitation28,29 and the SGT1−HSP90 interaction,Citation23 we propose that the SGT1-HSP90 complex is required for the localization of CENP-A to centromeres.
Yeast SGT1 (the homolog of SGT1/SUGT1 in humans) was originally identified not only as a dosage suppressor of the skp1–4 mutation, which causes defects in yeast kinetochore function, but also as a novel subunit of the Skp1-Cullin-F-box (SCF) ubiquitin ligase complex.Citation9 Therefore, we hypothesized that SKP1 is also involved in the CENP-A loading pathway. However, SKP1 siRNA treatment did not cause any signal reduction of CENP-A at centromeres (Fig. S3A and B; Table S2). Based on this result, we concluded that the SGT1-HSP90 complex participates in CENP-A deposition at centromeres in a SKP1-independent and/or SCF-independent manner. This conclusion is consistent with our previous report that the CUL4A-RBX1 complex, which does not require Skp1 to function, is required for CENP-A deposition at centromeres.Citation6
Altogether, our results indicate that specific downregulation of the endogenous SGT1-HSP90 complex in vivo leads to delocalization of CENP-A from centromeres; therefore, we conclude that the SGT1-HSP90 complex is specifically required to localize CENP-A to centromeres.
SGT1-HSP90 complex is required for CENP-A ubiquitylation in vivo
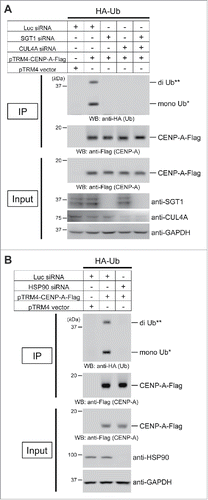
We previously reported that CENP-A K124 ubiquitylation, mediated by CUL4A-RBX1-COPS8 E3 ligase activity, is required for CENP-A deposition at the centromere.Citation6 As our results suggest that the SGT1-HSP90 complex is specifically required for the localization of CENP-A to centromeres (; Fig. S1 and S2), we next hypothesized that the SGT1-HSP90 complex is required for the ubiquitylation of CENP-A. To test this hypothesis, we performed in vivo ubiquitylation assays using HeLa Tet-Off cells harboring pcDNA3-HA-Ubiquitin plus pTRM4-CENP-A-Flag or pTRM4 vector only (see Materials and Methods).Citation6,30 We first observed that CENP-A–Flag proteins were ubiquitylated in HeLa cells (; compare 1st and 2nd lanes from the left), as previously reported.Citation6 Depletion of SGT1 significantly reduced these ubiquitylated forms (; compare 2nd and 3rd-5th lanes from the left; 4th lane is CUL4A siRNA control)Citation6 under the same conditions in which SGT1 siRNA abolished CENP-A centromeric localization (Fig. S1A). Further, we observed that HSP90 siRNA significantly reduced the ubiquitylation of CENP-A (). Taken together, these results support our hypothesis that the SGT1-HSP90 complex is required for CENP-A ubiquitylation.
Figure 2. The SGT1-HSP90 complex contributes to CENP-A monoubiquitylation and diubiquitylation in vivo. (A) Representative images of the in vivo ubiquitylation assay with the combination of SGT1 siRNA (#1 + #2), CUL4A siRNA (#1), or Luc siRNA control (Table S2; see CENP-A in vivo ubiquitylation assay in Materials and Methods). HeLa Tet-Off cells were cotransfected with the indicated constructs and siRNAs. Proteins in 5% of the total cell lysates (Input) and immunoprecipitates (IP) were detected by Western blot analysis using the indicated antibodies. GAPDH protein was the loading control (Input). (**) Putative di-Ub-CENP-A-Flag, (*) Putative mono-Ub-CENP-A-Flag. (B) Representative images of the in vivo ubiquitylation assay performed as (A) with the combination of HSP90 siRNA (#1) or Luc siRNA control (Table S2; see CENP-A in vivo ubiquitylation assay in Materials and Methods).
SGT1-HSP90 complex is required for composition of the CUL4A complex and recognition of CENP-A by COPS8
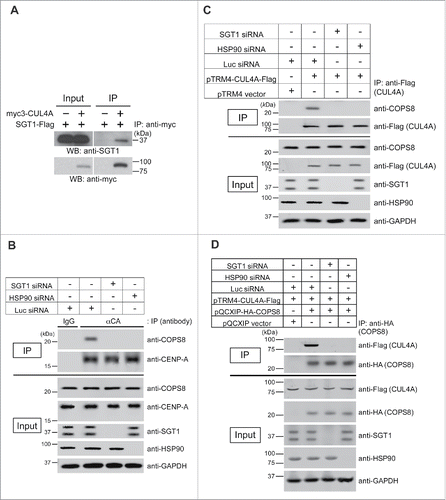
A question remains how the SGT1-HSP90 complex contributes to the E3 ligase activity of the CUL4A complex during CENP-A ubiquitylation. Previous studies, including our own, have indicated in vivo interactions of HSP90-CUL4ACitation31 and SGT1A-CUL4A (). We previously reported that the CUL4A complex uses the adaptor COPS8/CSN8 to target CENP-A.Citation6 Therefore, we hypothesized that the SGT1-HSP90 complex is important for the composition of the CUL4A complex and/or the recognition of CENP-A by COPS8. We tested whether depletion of SGT1 or HSP90 protein leads to loss of interaction among components of the CUL4A complex, including the adaptor protein COPS8 and the substrate CENP-A. Indeed, SGT1 or HSP90 siRNA abolished an interaction between COPS8 and CENP-A () and an interaction between COPS8 and CUL4A ( and ). These results suggest that the SGT1-HSP90 complex is required for composition of the CUL4A complex and recognition of CENP-A by COPS8.
Figure 3. SGT1-HSP90 complex is required for composition of the CUL4A complex and recognition of CENP-A by COPS8. (A) SGT1A interacts with CUL4A in vivo. Immunoblot analysis of CUL4A immunoprecipitates. Plasmids pTRM4-SGT1A-Flag and pcDNA3-myc3-CUL4A (Table S3) were cotransfected into HeLa Tet-Off cells. Forty-eight hours after transfection, protein extracts were prepared. Proteins in 1% of the total cell lysate (Input) and precipitate (IP) obtained by using anti-c-myc antibody (Table S1) were detected by Western blot analysis using anti-Flag antibody. (B) Interaction between CENP-A and COPS8 is reduced in response to SGT1 or HSP90 depletion. Immunoblot analysis of endogenous CENP-A immunoprecipitates. Indicated siRNA(s) (SGT1 #1 + #2; HSP90 #1; Luc; Table S2) were transfected into HeLa cells. Seventy-two hours after transfection, protein extracts were prepared. Proteins in 3% of the total cell lysate (Input) and precipitate (IP) obtained by using rabbit polyclonal anti-CENP-A antibody (Table S1) were detected by Western blot analysis using the indicated antibodies. GAPDH protein was the loading control (Input). (C) Interaction between CUL4A and COPS8 is reduced in response to SGT1 or HSP90 depletion. Immunoblot analysis of CUL4A immunoprecipitates. Plasmid pTRM4-CUL4A-Flag (Table S3) and indicated siRNA(s) (SGT1 #1 + #2; HSP90 #1; Luc; Table S2) were transfected into HeLa Tet-Off cells. Seventy-two hours after transfection, protein extracts were prepared. Proteins in 3% of the total cell lysate (Input) and precipitate (IP) obtained by using ANTI-FLAG M2 Affinity Gel (SIGMAALDRICH) were detected by Western blot analysis using the indicated antibodies. GAPDH protein was the loading control (Input). (D) Interaction between CUL4A and COPS8 is reduced in response to SGT1 or HSP90 depletion. Immunoblot analysis of COPS8 immunoprecipitates. The indicated plasmids (Table S3) and siRNA(s) (SGT1 #1 + #2; HSP90 #1; Luc; Table S2) were transfected into HeLa Tet-Off cells. Forty-eight hours after transfection, protein extracts were prepared. Proteins in 3% of the total cell lysate (Input) and precipitate (IP) obtained by using Anti-HA.11 Epitope Tag Affinity Matrix (Covance) were detected by Western blot analysis using the indicated antibodies. GAPDH protein was the loading control (Input).
SGT1 contributes to CENP-A deposition but not to the subsequent kinetochore assembly of central-outer kinetochore components
Davies et al. reported that because of its targeting of the MIS12 complex, the SGT1-HSP90 complex together with SKP1 is important for the accurate assembly of kinetochore protein complexes.Citation22 These investigators proposed that a balance between MIS12 complex assembly and turnover is required for the efficient and accurate assembly of kinetochore–microtubule-binding sites and that this balance is dependent on the SCF ubiquitin ligase subunit SKP1. Because our results suggest that SKP1 is not required for recruitment of CENP-A to centromeres, it is unlikely that SKP1 turnover affects the CENP-A loading pathway. In addition, because CENP-A is at the top of a hierarchy of the pathway that determines the assembly of kinetochore components,27 it is possible that destabilization of the MIS12 complex at the kinetochore observed by Davies et al. could be, at least in part, due to the defect in CENP-A recruitment.
To determine whether SGT1 contributes to kinetochore assembly of central-outer kinetochore components after CENP-A deposition at the centromere, we used the LacO/LacI ectopic centromeric chromatin assembly system. By this approach, we evaluated the ability of CENP-A fused to the Lac repressor (LacI) to recruit endogenous central-outer kinetochore components (MIS12, HEC1, and SKA1) at arrays of Lac operator (LacO) sequences on chromosome 1 of U2OS cells.Citation32,33 In this system, ectopic loci are forcibly determined through LacO-LacI interaction, and new centromeres can be generated artificially at ectopic sites through the assembly of LacI–CENP-A nucleosomes at LacO-containing arrays.Citation32,34,35 Our hypothetical scheme is the following: if SGT1 is required for CENP-A deposition at the centromere but not for downstream events such as the assembly of central-outer kinetochore components, then LacI–CENP-A expression will override defects due to SGT1 depletion and MIS12, HEC1, and SKA1 signals will not be affected at the ectopic loci of LacO arrays. If these signals are reduced at the centromere at the ectopic loci in LacO arrays, then we can conclude that SGT1 is required for kinetochore assembly of central-outer kinetochore components in addition to CENP-A deposition.
In prometaphase cells treated with SGT1 siRNA, we observed delocalization of HA-LacI-CENP-A at the native centromere (, green signal of samples [3] and [4], data not shown), a result that is consistent with our present data regarding endogenous CENP-A delocalization (; Fig. S1A). Depletion of SGT1 significantly reduced the ubiquitylation of HA-LacI-CENP-A (Fig. S4A), and this significant reduction is consistent with our present data regarding CENP-A ubiquitylation (). We also observed delocalization of endogenous MIS12, HEC1, and SKA1 proteins at the native kinetochore (, red signal of samples [3] and [4], data not shown) and abnormal chromosome congression; these observations are consistent with results of a previous reportCitation24 (, DAPI signal of samples [3] and [4], Fig. S4B). However, after ectopic loci were forcibly determined through LacO-LacI interaction, SGT1 siRNA treatment did not significantly affect the recruitment of endogenous MIS12, HEC1, and SKA1 proteins at LacO arrays ( and , green and red signals of insets; compare samples [2] and [4]). We confirmed that expression levels of HA-LacI-CENP-A and endogenous central-outer kinetochore proteins (MIS12, HEC1, and SKA1) are not affected in the cells treated with SGT1 siRNA (; compare samples [2] and [4]; only the HEC1 protein level was slightly increased by LacI-CENP-A expression but was unchanged between samples [2] and [4].). These results suggest that SGT1 contributes to CENP-A deposition but not to the subsequent kinetochore assembly of central-outer kinetochore components. Thus, SGT1 defects result not only in the reduction of immunofluorescence signals of the central-outer kinetochore proteins at the kinetochore, including ones reported previously,Citation22-24 but also in CENP-A mislocalization.
Figure 4. Lac-I-CENP-A expression overrides SGT1 depletion defects, and MIS12, HEC1, and SKA1 signals are not affected at the ectopic loci of LacO arrays. (A) Representative immunofluorescence images of MIS12, HEC1, and SKA1 recruitment to LacO arrays in prometaphase cells and the indicated HA-LacI-fused constructs with SGT1 siRNA (#1 + #2) or Luc siRNA control (Table S2) are shown. DAPI (blue), HA (green; low and high brightness), and endogenous MIS12, HEC1, or SKA1 (red) were visualized. Scale bar, 10 μm. Insets show magnification of the region of LacO arrays. (B) Histogram showing quantified MIS12, HEC1, and SKA1 fluorescent intensity at LacO arrays of the indicated HA-LacI constructs shown in (A) (ratio of the MIS12, HEC1, or SKA1 intensity to HA intensity at LacO arrays was normalized with sample [2]). More than 20 individual signals at LacO arrays in prometaphase cells were quantified, and the mean percentages ( ± SEM) are shown. *** P < 0.001 when sample [2] was compared, and not significant (n.s.) when the 2 indicated data sets were compared (Student's t test). (C) Expression levels of HA-LacI-CENP-A and endogenous central-outer kinetochore proteins (MIS12, HEC1, and SKA1) are not affected in the cells treated with SGT1 siRNA (#1 + #2) or Luc siRNA control (Table S2). U2OS-LacO cells were cultured and harvested 48 h after transfection with the indicated pcDNA3.1-HA-LacI constructs as shown in (A) and (B). Expression levels were confirmed by Western blot analysis with the indicated antibodies. GAPDH protein was used as a loading control.
![Figure 4. Lac-I-CENP-A expression overrides SGT1 depletion defects, and MIS12, HEC1, and SKA1 signals are not affected at the ectopic loci of LacO arrays. (A) Representative immunofluorescence images of MIS12, HEC1, and SKA1 recruitment to LacO arrays in prometaphase cells and the indicated HA-LacI-fused constructs with SGT1 siRNA (#1 + #2) or Luc siRNA control (Table S2) are shown. DAPI (blue), HA (green; low and high brightness), and endogenous MIS12, HEC1, or SKA1 (red) were visualized. Scale bar, 10 μm. Insets show magnification of the region of LacO arrays. (B) Histogram showing quantified MIS12, HEC1, and SKA1 fluorescent intensity at LacO arrays of the indicated HA-LacI constructs shown in (A) (ratio of the MIS12, HEC1, or SKA1 intensity to HA intensity at LacO arrays was normalized with sample [2]). More than 20 individual signals at LacO arrays in prometaphase cells were quantified, and the mean percentages ( ± SEM) are shown. *** P < 0.001 when sample [2] was compared, and not significant (n.s.) when the 2 indicated data sets were compared (Student's t test). (C) Expression levels of HA-LacI-CENP-A and endogenous central-outer kinetochore proteins (MIS12, HEC1, and SKA1) are not affected in the cells treated with SGT1 siRNA (#1 + #2) or Luc siRNA control (Table S2). U2OS-LacO cells were cultured and harvested 48 h after transfection with the indicated pcDNA3.1-HA-LacI constructs as shown in (A) and (B). Expression levels were confirmed by Western blot analysis with the indicated antibodies. GAPDH protein was used as a loading control.](/cms/asset/47477e09-ab0f-4ea9-9494-c6c211b53448/kccy_a_1325039_f0004_c.gif)
Discussion
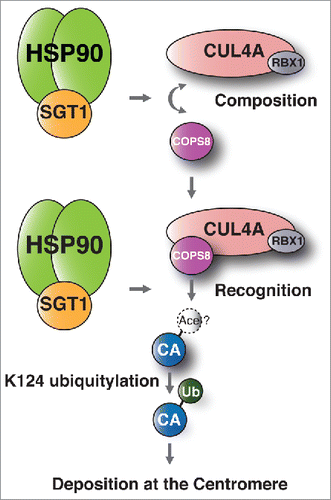
In the present study, we found that the SGT1-HSP90 complex is a positive regulator of CENP-A loading on centromeres. Based on this finding, we provide a model of the function of the HSP90-SGT1 complex in CENP-A deposition at the centromere (). The SGT1-HSP90 complex is required for CENP-A ubiquitylation () and localization of CENP-A to centromeres (; Fig. S1), because the SGT1-HSP90 complex is required for composition of the CUL4A complex and recognition of CENP-A by COPS8 (). Our results also suggest an interaction between SGT1 and CUL4A () and are in agreement with a previous finding that suggests an HSP90-CUL4A interaction.Citation31 In addition, our results clearly showed that SGT1 siRNA treatment did not significantly change the recruitment of endogenous MIS12, HEC1, and SKA1 proteins at LacO arrays, after ectopic loci were forcibly determined through LacO-LacI-CENP-A interaction ( and , green and red signals in insets; compare samples [2] and [4]). This lack of change in recruitment suggests that the SGT1 defect–induced reduction of immunofluorescence signals of the central-outer kinetochore proteins at the kinetochore, including ones reported previously,Citation22-24 are explained by CENP-A mislocalization caused by SGT1 defects.
Figure 5. Models of the functional contribution of the SGT1-HSP90 complex to CENP-A-deposition. The SGT1-HSP90 complex is required for composition of the CUL4A complex and recognition of COPS8 to target CENP-A. Therefore, the SGT1-HSP90 complex is required for CENP-A ubiquitylation and localization of CENP-A to centromeres. “CA” refers to the CENP-A monomer. “Ace” refers to the putative acetylated lysine 124 (K124) previously reported by Bui et al.Citation61
Liu et al. reported that PLK1 phosphorylates SGT1 at the kinetochore to promote timely kinetochore-microtubule attachment.Citation25 In their study, PLK1 was required for the prometaphase-specific localization of SGT1 to the kinetochore, and PLK1-mediated phosphorylation of SGT1 serine 331 (S331) at the kinetochore enhanced the association of HSP90-SGT1 chaperone with the MIS12 complex. This complex was stabilized at the kinetochore by the interaction and thus coordinates the recruitment of the NDC80 complex to form efficient microtubule-binding sites.Citation25 Combining the model proposed by Liu et al., we hypothesize that one possible pathway of the functional contribution of the SGT1-HSP90 complex is initiated by PLK1-mediated phosphorylation of SGT1 S331 (in humans) after CDK1-mediated priming phosphorylation of SGT1 S249 (this priming phosphorylation generates a docking site for the Polo-box domain of PLK1)Citation25 (Fig. S4C, G2/M and prometaphase at CEN). The timing of SGT1 delocalization at the kinetochore (i.e., metaphase) and dephosphorylation of S331 (i.e., anaphase) reported by Liu et al.Citation25 is slightly earlier than the timing of CENP-A deposition at the centromere (i.e., telophase/early G1) reported previously.Citation36 These results suggest that SGT1 phosphorylation is inhibitory at the specific time (i.e., telophase/early G1) of activation of the CUL4A complex. Proper dephosphorylation could be required subsequently (Fig. S4C, “dephosphorylation?”), although murine Sgt1-S302 (equivalent to human SGT1-S331) is thought to be required for the association of the Hsp90-Sgt1 chaperone with the MIS12 complex and proper assembly of the MIS12 complex at the kinetochores, but not for the kinetochore localization of murine Sgt1.Citation25 Taken together, each type of SGT1 phosphorylation may lead to prometaphase localization of SGT1 to the kinetochore, activation of the CUL4A complex, and subsequent ubiquitylation of CENP-A (Fig. S4C, M/G1 and M/G1 at CEN).
In addition to the stabilization of the MIS12 complex and deposition of CENP-A, other potential roles of the SGT1-HSP90 complex in kinetochore formation could not be ruled out by our results. McKinley et al. reported that in the G1 phase, PLK1 at centromeres binds to and phosphorylates the MIS18 complex to promote its localization and to license CENP-A deposition.Citation2 These investigators demonstrated that PLK1 localizes to G1 centromeres in a MIS18 complex-dependent manner, that PLK1 activity is required for new CENP-A deposition and the localization of the MIS18 complex and HJURP (but not CENP-C) to G1 centromeres.Citation2 Although the findings of McKinley et al suggest that PLK1 phosphorylation controls CENP-A deposition primarily by regulating M18BP1 localization, artificial targeting of M18BP1 to centromeres as a CENP-C-M18BP1Plk1-A fusion did not fully bypass recruitment for PLK1 activity, and they speculated that the phosphorylation sites of M18BP1 may regulate aspects of M18BP1 function in addition to controlling its localization.Citation2 Combining our findings with the model developed by McKinley et al., we propose that the SGT1-HSP90 complex contributes to another possible pathway, which presumably requires PLK1 and/or distinct CDK activity, to MIS18 complex recruitment and/or its phosphorylation and licensing process for CENP-A deposition in mammalian cells. Liu et al. also noted that dephosphorylation of S331 occurred slightly later than mitotic dephosphorylation of histone H3, which begins during anaphase, suggesting that dephosphorylation of SGT1 S331 probably occurs at the end of mitosisCitation25 (Fig. S4C, “dephosphorylation?”). Interestingly, this timing of dephosphorylation matches very well with the initiation of the full MIS18 complex localization, HJURP recruitment, and new CENP-A deposition at the centromere.Citation2,25,36-41 Further study is required to investigate how SGT1 dephosphorylation functionally correlates with the licensing process for CENP-A deposition by the MIS18 complex.Citation2,37,38,42
One controversial issue remains: why did not SGT1 or HSP90 siRNA or 17-AAG treatment in our previous studiesCitation23 and those of othersCitation24 affect the centromeric localization of CENP-C? Either treatment significantly affected the centromeric localization of CENP-A in the present study (; Fig. S1 and S2) and a hierarchical CENP-A–dependent CENP-C assembly mechanism was previously reported.Citation27 In human cells, 10% of the normal level of CENP-A is sufficient to drive kinetochore assemblyCitation27 and severe loss to less than 10% of the normal level of endogenous CENP-A is required to prevent endogenous CENP-C localization at centromeresCitation27,30; therefore, one possible explanation for the lack of effect on CENP-C localization is that SGT1 or HSP90 siRNA or 17-AAG treatment in our previous studyCitation23 and that of othersCitation24 was insufficient to reduce endogenous CENP-A to less than 10% of its normal level. Therefore, we performed a series of CENP-A siRNA at different concentrations (Fig. S5A; samples [1]-[5]) and immunofluorescence quantification of CENP-A, CENP-C, and CENP-I signals at centromeres. Indeed, we observed that a severe loss of endogenous CENP-A at the centromere (Figs. S5B and S5C; 5.2% of CENP-A in sample [5]) is required to prevent endogenous CENP-C localization at the centromere (Fig. S5B and C; 5.1% of CENP-C in sample [5]). In contrast, CENP-I was delocalized from the centromere substantially (Fig. S5B and C; 59.8% of CENP-I in sample [4]) when ∼20% of endogenous CENP-A (Fig. S5B and S5C) still remained at centromeres; the centromeric localization of CENP-C was not affected (Figs. S5B and S5C; 90.7% of CENP-C in sample [4]). This sample [4] result mimics the phenotypes seen with SGT1 or HSP90 siRNA (; Fig. S1) or 17-AAG treatment (Fig. S2), which did not affect the centromeric localization of CENP-C. The number of abnormal metaphase cells was highly associated with the degrees of CENP-A depletion (Fig. S6A and B, CENP-A [2]-[5]) and delocalization from centromeres (Fig. S5); this association is comparable to ones in previous reportsCitation23,43 (Note that 25–30% misaligned metaphase cells were previously observed with 17-AAG treatment [500 nM for 24 h]; this proportion is similar to that seen with the present sample [4],23 and 67% of misaligned metaphase cells were reported with CENP-A siRNA; this proportion is comparable to that observed for current sample [5]Citation43). In addition, recent works suggested that CENP-C serves as a centromere-localized binding partner for the MIS18 complex: CENP-C is required for MIS18 complex recruitment to centromeres to promote CENP-A chromatin assembly.Citation2,27,44 These studies suggested a dual function of CENP-C before and after CENP-A deposition at the centromere. In addition, it is possible that a CENP-A–independent CENP-C assembly mechanism exists at the centromere, especially during the time of MIS18 complex recruitment.
Ectopic incorporation of overexpressed CENP-A can result in genomic instability,45-48 which occurs in particularly aggressive cancer cells and tissues.Citation49-53 In humans, CENP-A overexpression can lead to its ectopic localization to chromosome regions with active histone turnover, as seen in cancer cell linesCitation49,54; therefore, CENP-A is a reasonable target for cancer therapy. Cancer cells also utilize HSP90 as a chaperone to promote folding and function of mutated or overexpressed oncoproteins, because aberrant oncoproteins are unstable.Citation15,21 SGT1 is also involved in human cancer development by stabilizing proteins that are required for growth of cancer cells.Citation21 Therefore, the SGT1-HSP90 complex is a potential target for cancer therapy. It would be interesting to study how effective and specific combinational therapies directed at these targets could be.
Materials and methods
Antibodies and reagents
Please see Table S1 for antibodies, Table S2 for siRNA sequences, and Table S3 for plasmid vectors used in this study.
Cell culture and transfection
HeLa or HeLa Tet-Off cells (Clontech, Mountain View, CA) were cultured in high-glucose Dulbecco's modified Eagle's medium (BioWhittaker, Walkersville, MD) with 10% fetal bovine serum (FBS) (Invitrogen, Grand Island, NY). Cells were grown at 37°C in 5% CO2 in a humidified incubator. Cells were transfected with annealed double-stranded siRNA(s) and/or mammalian expression plasmids by using Lipofectamine 2000 (Invitrogen), Lipofectamine 3000 (Invitrogen), Lipofectamine LTX (Invitrogen), Lipofectamine RNAiMAX (Invitrogen), or linear polyethylenimine (PEI).Citation55 HeLa Tet-Off cells were cultured without tetracycline/doxycycline and transiently transfected with the pTRM4 overexpression vector whose transcription was regulated by the TRE promoter. 17-AAG treatment was performed as described previously.Citation56,57
Immunoblotting
The method of immunoblotting has been described in detail previously.Citation6,23,26,56,57 The Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE), the Odyssey Fc Infrared imaging System (LI-COR Biosciences, Lincoln, NE), the Odyssey CLx Infrared imaging System (LI-COR Biosciences, Lincoln, NE), Molecular Imager Versadoc MP4000 System (Bio-Rad, Hercules, CA), and/or FluorChem M system (ProteimSimple, San Jose, CA) were used for detection of coimmunoblotting. Cells were suspended in denaturing buffer A1 (20 mM Tris-HCl pH 7.4, 50 mM NaCl, 0.5% Nonidet P-40, 0.5% deoxycholate, 0.5% SDS, 1 mM EDTA, and complete EDTA-free protease inhibitor cocktail [Roche, Indianapolis, IN, USA]),Citation6 buffer A2 (20 mM Tris-HCl pH 7.4, 50 mM NaCl, 0.5% Nonidet P-40, 0.5% deoxycholate, 0.5% SDS, 1 mM EDTA, 50 μM MG132, and complete EDTA-free protease inhibitor cocktail [Roche]),Citation58 or buffer used in the immunoprecipitation assay. The cell suspension was sonicated, frozen in liquid nitrogen, and thawed (freeze-thaw process). Before electrophoresis, cell lysates were mixed with SDS sample buffer.Citation59 Proteins were subjected to Western blot analysisCitation23,56,57 with the indicated antibodies (Table S1). Anti-CENP-C rabbit serum (serum 554) was generously provided by Dr. William C. Earnshaw (Wellcome Trust Centre for Cell Biology, University of Edinburgh, UK; Table S1). The intensity of band signals was quantified by Quantity One 1-D Analysis Software Version 4.6.9 (Bio-Rad, Hercules, CA), Image Studio Version 5.2 Software (LI-COR Biosciences, Lincoln, NE), and/or ImageJ 1.51m9 (NIH, Bethesda, MD).
Immunoprecipitation assay
The immunoprecipitation assay was performed as described previouslyCitation6,23,26,56,57 with the following minor modifications. HeLa Tet-Off cells were cultured without tetracycline/doxycycline and transfected with the indicated plasmids (Table S3).
To study the interaction of SGT1A-Flag with myc3-CUL4A(), HeLa Tet-Off cells were collected at 48 h after transfection, washed with PBS, and lysed in ice-cold buffer B (50 mM HEPES, pH 7.2, 120 mM NaCl, 0.1% NP-40, 1 mM EDTA and complete EDTA-free protease inhibitor cocktail [Roche]) for 30 min at 4°C. After lysates underwent centrifugation (21,000 g for 30 min at 4°C), proteins were immunoprecipitated overnight with c-Myc Monoclonal Antibody-Agarose Beads (Clontech, Mountain View, CA, USA) pre-blocked with 1% BSA in PBS. Immunoprecipitates were washed 3 times with buffer B, and proteins were eluted with SDS sample bufferCitation59 and subjected to Western blot analysisCitation23,56,57with the indicated antibodies.
To study the interaction of endogenous CENP-A with endogenous COPS8 (), HeLa cells were transfected with pCGN-HA-Ubiquitin (Table S3). At 48 h after transfection, cells were incubated with 5 μM MG132 (CALBIOCHEM) for 24 h (this condition mimics that used in the CENP-A in vivo ubiquitylation assay). Cells were collected at 72 h after transfection and lysed in buffer C (100 mM Tris-HCl pH 7.5, 500 mM NaCl, 0.5% Triton X-100, 0.05% Nonidet P-40, 0.1% Tween 20, 0.1% [w/v] CHAPS, and complete EDTA-free protease inhibitor cocktail [Roche]) by a sonication and freeze-thaw process. After lysates underwent centrifugation (10,000 g for 3 min at 4°C), proteins were immunoprecipitated overnight at 4°C in buffer C; for immunoprecipitation we used Protein A Sepharose CL-4B (GE Healthcare, Pittsburgh, PA) pre-incubated overnight at 4°C with anti-CENP-A rabbit polyclonal antibody (αCA) (Upstate, Table S1) or normal rabbit IgG control (IgG) (Santa Cruz, Table S1) in buffer C. Immunoprecipitates were washed 4 times with buffer C, and proteins were eluted with SDS sample bufferCitation59 and subjected to Western blot analysisCitation23,56,57 with the indicated antibodies (Table S1).
To study the interaction of exogenous CUL4A-Flag with endogenous COPS8 (), HeLa Tet-Off cells were cotransfected with plasmids pTRM4-CUL4A-Flag and pCGN-HA-Ubiquitin (Table S3; see Cell culture and transfection). At 48 h after transfection, cells were incubated with 5 μM MG132 (CALBIOCHEM) for 24 h (this condition mimics that used in the CENP-A in vivo ubiquitylation assay). Cells were collected at 72 h after transfection and lysed in buffer C (100 mM Tris-HCl pH 7.5, 500 mM NaCl, 0.5% Triton X-100, 0.05% Nonidet P-40, 0.1% Tween 20, 0.1% [w/v] CHAPS, and complete EDTA-free protease inhibitor cocktail [Roche]) by a sonication and freeze-thaw process. After lysates underwent centrifugation (10,000 g for 3 min at 4°C), proteins were immunoprecipitated with anti-Flag M2 affinity gel (SIGMA-ALDRICH) overnight at 4°C in buffer C. Immunoprecipitates were washed 4 times with buffer C, and proteins were eluted with SDS sample bufferCitation59 and subjected to Western blot analysisCitation23,56,57 with the indicated antibodies (Table S1).
To study the interaction of exogenous CUL4A-Flag with exogenous HA-COPS8 (), we subjected HeLa Tet-Off cells to cotransfection with plasmids pTRM4-CUL4A-Flag and pQCXIP-HA-COPS8 (Table S3; see Cell culture and transfection) and incubated them for 48 h. Cells were collected and lysed in buffer C (100 mM Tris-HCl pH 7.5, 500 mM NaCl, 0.5% Triton X-100, 0.05% Nonidet P-40, 0.1% Tween 20, 0.1% [w/v] CHAPS, and complete EDTA-free protease inhibitor cocktail [Roche]) by a sonication and freeze-thaw process. After lysates underwent centrifugation (10,000 g for 3 min at 4°C), proteins were immunoprecipitated with Anti-HA.11 Epitope Tag Affinity Matrix (Covance) for 3 h at 4°C in buffer C. Immunoprecipitates were washed 4 times with buffer C, and proteins were eluted with SDS sample bufferCitation59 and subjected to Western blot analysisCitation23,56,57 with the indicated antibodies.
Immunofluorescence
The indirect immunofluorescence staining was performed as described previouslyCitation6,23,26,30,56,57,60 with the following minor modifications. To detect endogenous CENP-A proteins in HeLa or HeLa Tet-Off cells, approximately 5.4 × 105 cells per well were seeded on cover glasses (22 mm x 22 mm) that were placed in a 6-well polystyrene plate. Cells were cultured without tetracycline/doxycycline, grown for 18 h before transfection, and fixed at 48 h or 72 h after transfection with the indicated siRNA (Table S2).
To detect endogenous CENP-A with 17-AAG treatment (Fig. S2A-C), approximately 1.8 to 5.4 × 105 HeLa or HeLa Tet-Off cells per well were seeded on cover glasses (22 mm × 22 mm) that were placed in a 6-well polystyrene plate, and cells were grown for 18 h before 17-AAG was added. Cells were incubated with various concentrations of 17-AAG for 24 h as indicated (Fig. S2A-C).
Image acquisition and processing, including deconvolution and signal quantitation, were performed by Openlab version 5.5.2 Scientific Imaging Software (Improvision, Lexington, MA) or Velocity version 6.3 3D Image Analysis Software (Improvision, Lexington, MA). We used CENP-B signals as reference signals for all data.
CENP-A In Vivo ubiquitylation assay
The in vivo CENP-A ubiquitylation assay was performed as described previouslyCitation6 with the following minor modifications.
In the study of ubiquitylation of exogenous CENP-A-Flag ( and ), HeLa Tet-Off cells were cotransfected with the indicated expression vectors (Table S3) and siRNA(s) (Table S2) plus pCGN-HA-Ubiquitin (Table S3; see Cell culture and transfection). At 24 h () or 48 h () after transfection, cells were incubated with 5 μM MG132 (CALBIOCHEM) for 24 h. Cells were collected at 48 h () or 72 h () after transfection and lysed in denaturing buffer A2 (20 mM Tris-HCl pH 7.4, 50 mM NaCl, 0.5% Nonidet P-40, 0.5% deoxycholate, 0.5% SDS, 1 mM EDTA, 50 μM MG132, and complete EDTA-free protease inhibitor cocktail [Roche])Citation6 by a sonication and freeze-thaw process. Proteins were immunoprecipitated with anti-Flag M2 affinity gel (SIGMA-ALDRICH) overnight at 4°C in buffer A2, and the immunoprecipitates underwent Western blot analysis with the indicated antibodies.
For the study of ubiquitylation of exogenous HA-LacI-CENP-A (Fig. S4A), plasmids pcDNA3.1-HA-LacI, pcDNA3.1-HA-LacI-CENP-A, and U2OS-LacO cells were generously provided by Dr. Ben E. Black (Department of Biochemistry and Biophysics, Perelman School of Medicine, University of Pennsylvania; Table S3). The plasmid pcDNA3–6xHis-Ubiquitin was a generous gift from Dr. Dawn S. Chandler (The Research Institute at Nationwide Children's Hospital; Table S3). U2OS-LacO cells were cotransfected with the indicated expression vectors (Table S3) and siRNA(s) (Table S2) plus pcDNA3–6xHis-Ubiquitin (Table S3; see Cell culture and transfection) by using Lipofectamine 3000 (Invitrogen). At 24 h after transfection, cells were incubated with 5 μM MG132 (CALBIOCHEM) for 24 h. Cells were collected at 48 h after transfection and lysed in denaturing buffer A2 by a sonication and freeze-thaw process. Proteins were immunoprecipitated with Anti-HA.11 Epitope Tag Affinity Matrix (Covance) overnight at 4°C in buffer A2, and the immunoprecipitates underwent Western blot analysis with the indicated antibodies.
LacO/LacI ectopic centromeric chromatin assembly system
Plasmids pcDNA3.1-HA-LacI, pcDNA3.1-HA-LacI-CENP-A, and U2OS-LacO cells were generously provided by Dr. Ben E. Black (Department of Biochemistry and Biophysics, Perelman School of Medicine, University of Pennsylvania; Table S3). The LacO/LacI ectopic centromeric chromatin assembly was performed as described previouslyCitation30 with the following minor modifications. Approximately 4.8 × 104 U2OS-LacO cells per chamber (0.7 cm2) were seeded in 8-chamber polystyrene vessels (tissue culture-treated glass slides; BD Falcon), and cells were grown for 18 h before transfection. Cells were transfected with the indicated construct(s) (; Fig. S4B) by using Lipofectamine 3000 (Invitrogen) or linear polyethylenimine (PEI)Citation55 and grown for 48 h on coverslip slides. For MIS12, HEC1, or SKA1 staining, samples were fixed and stained as described previously.Citation30 For MIS12 or SKA1 staining, samples were fixed with 4% paraformaldehyde solution at 4°C and subsequently stained as described previously,Citation60 but all the other processes were performed at room temperature (ca. 25°C). Dilution of primary antibodies were as follow (Table S1): anti-HA mouse monoclonal antibody (Roche), 1/100; anti-HA rabbit polyclonal antibody (Santa Cruz), 1/10; anti-MIS12 rabbit polyclonal antibody (abcam), 1/100; anti-HEC1 mouse monoclonal antibody (Novus/GeneTex), 1/100; anti-SKA1 rabbit polyclonal antibody (abcam), 1/100. For cell counting to evaluate abnormal chromosome congression (Fig. S4B), cells were stained with anti-phospho-histone H3 mouse monoclonal antibody (abcam; dilution ratio: 1/400; Table S1). Image acquisition and quantitation were performed as described previously.Citation6,30,60
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Author contributions
YN, Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting or revising the article; RK, Contributed to construction; HO, Acquisition of data; KK, Conception and design, Drafting or revising the article.
1325039__Supplemental_Material.pdf
Download PDF (4.2 MB)Acknowledgments
The authors thank Ben E. Black, Glennis A. Logsdon, Dawn S. Chandler, and William C. Earnshaw for their generous gifts of reagents. The authors thank past and current researchers at The Research Institute at Nationwide Children's Hospital and St. Jude Children's Research Hospital for their helpful discussions, experimental guidance, and reagents.
Funding
This study was supported by NIH grant CA205659.
Related Research Data
References
- Black BE, Jansen LE, Foltz DR, Cleveland DW. Centromere identity, function, and epigenetic propagation across cell divisions. Cold Spring Harb Symp Quant Biol 2010; 75:403-18; PMID:21467140; https://doi.org/10.1101/sqb.2010.75.038
- McKinley KL, Cheeseman IM. Polo-like kinase 1 licenses CENP-A deposition at centromeres. Cell 2014; 158:397-411; PMID:25036634; https://doi.org/10.1016/j.cell.2014.06.016
- Warburton PE, Cooke CA, Bourassa S, Vafa O, Sullivan BA, Stetten G, Gimelli G, Warburton D, Tyler-Smith C, Sullivan KF, et al. Immunolocalization of CENP-A suggests a distinct nucleosome structure at the inner kinetochore plate of active centromeres. Curr Biol 1997; 7:901-4; PMID:9382805; https://doi.org/10.1016/S0960-9822(06)00382-4
- Vissel B, Choo KH. Human alpha satellite DNA–consensus sequence and conserved regions. Nucleic Acids Res 1987; 15:6751-2; PMID:3628014; https://doi.org/10.1093/nar/15.16.6751
- Fukagawa T, Earnshaw WC. The centromere: chromatin foundation for the kinetochore machinery. Dev Cell 2014; 30:496-508; PMID:25203206; https://doi.org/10.1016/j.devcel.2014.08.016
- Niikura Y, Kitagawa R, Ogi H, Abdulle R, Pagala V, Kitagawa K. CENP-A K124 Ubiquitylation Is Required for CENP-A Deposition at the Centromere. Dev Cell 2015; 32:589-603; PMID:25727006; https://doi.org/10.1016/j.devcel.2015.01.024
- Rago F, Gascoigne KE, Cheeseman IM. Distinct organization and regulation of the outer kinetochore KMN network downstream of CENP-C and CENP-T. Curr Biol 2015; 25:671-7; PMID:25660545; https://doi.org/10.1016/j.cub.2015.01.059
- Hori T, Shang WH, Takeuchi K, Fukagawa T. The CCAN recruits CENP-A to the centromere and forms the structural core for kinetochore assembly. J Cell Biol 2013; 200:45-60; PMID:23277427; https://doi.org/10.1083/jcb.201210106
- Kitagawa K, Skowyra D, Elledge SJ, Harper JW, Hieter P. SGT1 encodes an essential component of the yeast kinetochore assembly pathway and a novel subunit of the SCF ubiquitin ligase complex. Mol Cell 1999; 4:21-33; PMID:10445024; https://doi.org/10.1016/S1097-2765(00)80184-7
- Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p-mediated phosphorylation. Cell 1997; 91:491-500; PMID:9390558; https://doi.org/10.1016/S0092-8674(00)80435-3
- Stemmann O, Neidig A, Kocher T, Wilm M, Lechner J. Hsp90 enables Ctf13p/Skp1p to nucleate the budding yeast kinetochore. Proc Natl Acad Sci U S A 2002; 99:8585-90; PMID:12084919; https://doi.org/10.1073/pnas.082223899
- Goetz MP, Toft DO, Ames MM, Erlichman C. The Hsp90 chaperone complex as a novel target for cancer therapy. Ann Oncol 2003; 14:1169-76; PMID:12881371; https://doi.org/10.1093/annonc/mdg316
- Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol 2003; 15:419-24; PMID:14624223; https://doi.org/10.1097/00001622-200311000-00003
- Workman P. Overview: translating Hsp90 biology into Hsp90 drugs. Curr Cancer Drug Targets 2003; 3:297-300; PMID:14529382; https://doi.org/10.2174/1568009033481868
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 2010; 10:537-49; PMID:20651736; https://doi.org/10.1038/nrc2887
- Bansal PK, Abdulle R, Kitagawa K. Sgt1 associates with Hsp90: an initial step of assembly of the core kinetochore complex. Mol Cell Biol 2004; 24:8069-79; PMID:15340069; https://doi.org/10.1128/MCB.24.18.8069-8079.2004
- Hubert DA, Tornero P, Belkhadir Y, Krishna P, Takahashi A, Shirasu K, Dangl JL. Cytosolic HSP90 associates with and modulates the Arabidopsis RPM1 disease resistance protein. EMBO J 2003; 22:5679-89; PMID:14592967; https://doi.org/10.1093/emboj/cdg547
- Lee YT, Jacob J, Michowski W, Nowotny M, Kuznicki J, Chazin WJ. Human Sgt1 binds HSP90 through the CHORD-Sgt1 domain and not the tetratricopeptide repeat domain. J Biol Chem 2004; 279:16511-7; PMID:14761955; https://doi.org/10.1074/jbc.M400215200
- Lingelbach LB, Kaplan KB. The interaction between Sgt1p and Skp1p is regulated by HSP90 chaperones and is required for proper CBF3 assembly. Mol Cell Biol 2004; 24:8938-50; PMID:15456868; https://doi.org/10.1128/MCB.24.20.8938-8950.2004
- Takahashi A, Casais C, Ichimura K, Shirasu K. HSP90 interacts with RAR1 and SGT1 and is essential for RPS2-mediated disease resistance in Arabidopsis. Proc Natl Acad Sci U S A 2003; 100:11777-82; PMID:14504384; https://doi.org/10.1073/pnas.2033934100
- Ogi H, Sakuraba Y, Kitagawa R, Xiao L, Shen C, Cynthia MA, Ohta S, Arnold MA, Ramirez N, Houghton PJ, et al. The oncogenic role of the cochaperone Sgt1. Oncogenesis 2015; 4:e149; PMID:25985210; https://doi.org/10.1038/oncsis.2015.12
- Davies AE, Kaplan KB. Hsp90-Sgt1 and Skp1 target human Mis12 complexes to ensure efficient formation of kinetochore-microtubule binding sites. J Cell Biol 2010; 189:261-74; PMID:20404110; https://doi.org/10.1083/jcb.200910036
- Niikura Y, Ohta S, Vandenbeldt KJ, Abdulle R, McEwen BF, Kitagawa K. 17-AAG, an Hsp90 inhibitor, causes kinetochore defects: a novel mechanism by which 17-AAG inhibits cell proliferation. Oncogene 2006; 25:4133-46; PMID:16501598; https://doi.org/10.1038/sj.onc.1209461
- Steensgaard P, Garre M, Muradore I, Transidico P, Nigg EA, Kitagawa K, Earnshaw WC, Faretta M, Musacchio A. Sgt1 is required for human kinetochore assembly. EMBO Rep 2004; 5:626-31; PMID:15133482; https://doi.org/10.1038/sj.embor.7400154
- Liu XS, Song B, Tang J, Liu W, Kuang S, Liu X. Plk1 phosphorylates Sgt1 at the kinetochores to promote timely kinetochore-microtubule attachment. Mol Cell Biol 2012; 32:4053-67; PMID:22869522; https://doi.org/10.1128/MCB.00516-12
- Niikura Y, Kitagawa K. Identification of a novel splice variant: human SGT1B (SUGT1B). DNA Seq 2003; 14:436-41; PMID:15018354; https://doi.org/10.1080/10425170310001623644
- Liu ST, Rattner JB, Jablonski SA, Yen TJ. Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J Cell Biol 2006; 175:41-53; PMID:17030981; https://doi.org/10.1083/jcb.200606020
- Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003; 3:213-7; PMID:12676580; https://doi.org/10.1016/S1535-6108(03)00029-1
- Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther 2004; 3:1021-30; PMID:15299085; https://doi.org/10.4161/cbt.3.10.1142
- Niikura Y, Kitagawa R, Kitagawa K. CENP-A Ubiquitylation Is Inherited through Dimerization between Cell Divisions. Cell Rep 2016; 15:61-76; PMID:27052173; https://doi.org/10.1016/j.celrep.2016.03.010
- Dias DC, Dolios G, Wang R, Pan ZQ. CUL7: A DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc Natl Acad Sci U S A 2002; 99:16601-6; PMID:12481031; https://doi.org/10.1073/pnas.252646399
- Logsdon GA, Barrey EJ, Bassett EA, DeNizio JE, Guo LY, Panchenko T, Dawicki-McKenna JM, Heun P, Black BE. Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. J Cell Biol 2015; 208:521-31; PMID:25713413; https://doi.org/10.1083/jcb.201412011
- Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, et al. From silencing to gene expression: real-time analysis in single cells. Cell 2004; 116:683-98; PMID:15006351; https://doi.org/10.1016/S0092-8674(04)00171-0
- Barnhart MC, Kuich PH, Stellfox ME, Ward JA, Bassett EA, Black BE, Foltz DR. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J Cell Biol 2011; 194:229-43; PMID:21768289; https://doi.org/10.1083/jcb.201012017
- Mendiburo MJ, Padeken J, Fulop S, Schepers A, Heun P. Drosophila CENH3 is sufficient for centromere formation. Science 2011; 334:686-90; PMID:22053052; https://doi.org/10.1126/science.1206880
- Jansen LE, Black BE, Foltz DR, Cleveland DW. Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol 2007; 176:795-805; PMID:17339380; https://doi.org/10.1083/jcb.200701066
- Fujita Y, Hayashi T, Kiyomitsu T, Toyoda Y, Kokubu A, Obuse C, Yanagida M. Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev Cell 2007; 12:17-30; PMID:17199038; https://doi.org/10.1016/j.devcel.2006.11.002
- Hayashi T, Fujita Y, Iwasaki O, Adachi Y, Takahashi K, Yanagida M. Mis16 and Mis18 are required for CENP-A loading and histone deacetylation at centromeres. Cell 2004; 118:715-29; PMID:15369671; https://doi.org/10.1016/j.cell.2004.09.002
- Maddox PS, Hyndman F, Monen J, Oegema K, Desai A. Functional genomics identifies a Myb domain-containing protein family required for assembly of CENP-A chromatin. J Cell Biol 2007; 176:757-63; PMID:17339379; https://doi.org/10.1083/jcb.200701065
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 2009; 137:485-97; PMID:19410545; https://doi.org/10.1016/j.cell.2009.02.040
- Foltz DR, Jansen LE, Bailey AO, Yates JR, Bassett 3rd, EA, Wood S, Black BE, Cleveland DW. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell 2009; 137:472-84; PMID:19410544; https://doi.org/10.1016/j.cell.2009.02.039
- Kim IS, Lee M, Park KC, Jeon Y, Park JH, Hwang EJ, Jeon TI, Ko S, Lee H, Baek SH, et al. Roles of Mis18alpha in Epigenetic Regulation of Centromeric Chromatin and CENP-A Loading. Mol Cell 2012; 46:260-73; PMID:22516971; https://doi.org/10.1016/j.molcel.2012.03.021
- Goshima G, Kiyomitsu T, Yoda K, Yanagida M. Human centromere chromatin protein hMis12, essential for equal segregation, is independent of CENP-A loading pathway. J Cell Biol 2003; 160:25-39; PMID:12515822; https://doi.org/10.1083/jcb.200210005
- Dambacher S, Deng W, Hahn M, Sadic D, Frohlich J, Nuber A, Hoischen C, Diekmann S, Leonhardt H, Schotta G. CENP-C facilitates the recruitment of M18BP1 to centromeric chromatin. Nucleus 2012; 3:101-10; PMID:22540025; https://doi.org/10.4161/nucl.18955
- Amato A, Schillaci T, Lentini L, Di Leonardo A. CENPA overexpression promotes genome instability in pRb-depleted human cells. Mol Cancer 2009; 8:119; PMID:20003272; https://doi.org/10.1186/1476-4598-8-119
- Au WC, Crisp MJ, DeLuca SZ, Rando OJ, Basrai MA. Altered dosage and mislocalization of histone H3 and Cse4p lead to chromosome loss in Saccharomyces cerevisiae. Genetics 2008; 179:263-75; PMID:18458100; https://doi.org/10.1534/genetics.108.088518
- Heun P, Erhardt S, Blower MD, Weiss S, Skora AD, Karpen GH. Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Dev Cell 2006; 10:303-15; PMID:16516834; https://doi.org/10.1016/j.devcel.2006.01.014
- Van Hooser AA, Ouspenski II, Gregson HC, Starr DA, Yen TJ, Goldberg ML, Yokomori K, Earnshaw WC, Sullivan KF, Brinkley BR. Specification of kinetochore-forming chromatin by the histone H3 variant CENP-A. J Cell Sci 2001; 114:3529-42. PMID:11682612
- Tomonaga T, Matsushita K, Yamaguchi S, Oohashi T, Shimada H, Ochiai T, Yoda K, Nomura F. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res 2003; 63:3511-6. PMID:12839935
- Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF, Bernstein E. Histone variants: emerging players in cancer biology. Cell Mol Life Sci 2014; 71:379-404; PMID:23652611; https://doi.org/10.1007/s00018-013-1343-z
- Biermann K, Heukamp LC, Steger K, Zhou H, Franke FE, Guetgemann I, Sonnack V, Brehm R, Berg J, Bastian PJ, et al. Gene expression profiling identifies new biological markers of neoplastic germ cells. Anticancer Res 2007; 27:3091-100; PMID:17970049
- Wu Q, Qian YM, Zhao XL, Wang SM, Feng XJ, Chen XF, Zhang SH. Expression and prognostic significance of centromere protein A in human lung adenocarcinoma. Lung Cancer 2012; 77:407-14; PMID:22542705; https://doi.org/10.1016/j.lungcan.2012.04.007
- McGovern SL, Qi Y, Pusztai L, Symmans WF, Buchholz TA. Centromere protein-A, an essential centromere protein, is a prognostic marker for relapse in estrogen receptor-positive breast cancer. Breast Cancer Res 2012; 14:R72; PMID:22559056; https://doi.org/10.1186/bcr3181
- Lacoste N, Woolfe A, Tachiwana H, Garea AV, Barth T, Cantaloube S, Kurumizaka H, Imhof A, Almouzni G. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol Cell 2014; 53:631-44; PMID:24530302; https://doi.org/10.1016/j.molcel.2014.01.018
- Reed SE, Staley EM, Mayginnes JP, Pintel DJ, Tullis GE. Transfection of mammalian cells using linear polyethylenimine is a simple and effective means of producing recombinant adeno-associated virus vectors. J Virol Methods 2006; 138:85-98; PMID:16950522; https://doi.org/10.1016/j.jviromet.2006.07.024
- Niikura Y, Dixit A, Scott R, Perkins G, Kitagawa K. BUB1 mediation of caspase-independent mitotic death determines cell fate. J Cell Biol 2007; 178:283-96; PMID:17620410; https://doi.org/10.1083/jcb.200702134
- Niikura Y, Ogi H, Kikuchi K, Kitagawa K. BUB3 that dissociates from BUB1 activates caspase-independent mitotic death (CIMD). Cell Death Differ 2010; 17:1011-24; PMID:20057499; https://doi.org/10.1038/cdd.2009.207
- Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell 2006; 22:383-94; PMID:16678110; https://doi.org/10.1016/j.molcel.2006.03.035
- Lamb JR, Tugendreich S, Hieter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem Sci 1995; 20:257-9; PMID:7667876; https://doi.org/10.1016/S0968-0004(00)89037-4
- Niikura Y, Kitagawa K. Immunofluorescence Analysis of Endogenous and Exogenous Centromere-kinetochore Proteins. J Vis Exp 2016; PMID:26967065; https://doi.org/10.3791/53732
- Bui M, Dimitriadis EK, Hoischen C, An E, Quenet D, Giebe S, Nita-Lazar A, Diekmann S, Dalal Y. Cell-cycle-dependent structural transitions in the human CENP-A nucleosome in vivo. Cell 2012; 150:317-26; PMID:22817894; https://doi.org/10.1016/j.cell.2012.05.035