
53BP1 is a central player in the repair of DNA-double strand breaks (DSBs) as it orchestrates the choice between the DSB repair pathways, non-homologous end-joining (NHEJ) and homologous recombination (HR).Citation1 53BP1 promotes repair by NHEJ by countering the function of BRCA1 in the HR pathway. This antagonistic relationship of BRCA1 and 53BP1 has been revealed in the context of BRCA1 deficiency.Citation2 BRCA1-mutant cells are deficient in HR-mediated DNA repair and exquisitely sensitive to treatment with Poly-ADP-Ribose Polymerase (PARP) inhibitors. Loss of 53BP1 almost completely abolishes the sensitivity of BRCA1-mutant cells to PARP inhibitors, which also correlates with the restoration of competent HR. This finding illustrates the fact that 53BP1 and BRCA1 compete to repair the same DSB. In a physiologic scenario, the BRCA1 and 53BP1 interplay ensures that the repair is accurate and restricted to the appropriate cellular contexts.
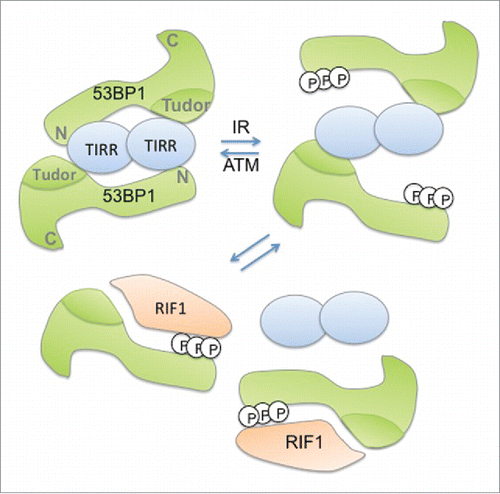
The newly identified 53BP1-partner TIRR represents a pathway that modulates DNA repair by restricting the access of 53BP1 to DNA lesions.Citation3,4 53BP1 and TIRR form a stable nuclear soluble complex in undamaged cells. Following DNA damage, TIRR dissociates from 53BP1 through a mechanism involving the phosphorylation of the N-terminal of 53BP1 by the Ataxia Telangiectasia Mutated (ATM) kinase and the recruitment of Rap1-interacting factor 1 homolog (RIF-1). This dissociation allows nuclear soluble 53BP1 to localize to chromatin. At the molecular level, TIRR interacts with the Tudor domain of 53BP1. This domain is involved in 53BP1 recruitment to the damaged chromatin by recognition of histone H4 dimethylated in lysine K20 (H4K20me2).Citation5 Structural evidence indicates that H4K20me2 and TIRR binding surfaces on 53BP1 Tudor domain overlap. However, the binding affinity of TIRR with Tudor is ∼25-fold stronger than the interaction of Tudor with H4K20me2 peptide.Citation3 Intriguingly the dissociation of TIRR from 53BP1 appears to involve the N-terminal region and not the Tudor region. The in vitro and structural studies were restricted to the Tudor domains of 53BP1 therefore it is feasible that the N-terminal end of 53BP1 also directly interacts with TIRR (). The disruption of this interaction via the recruitment of RIF-1 to 53BP1 is sufficient for the release of TIRR.
Figure 1. Speculative model depicting the TIRR/53BP1 dissociation in response to γ-radiation. In undamaged cells, 2 53BP1 molecules interact with a dimer of TIRR via their Tudor domain and possibly their N-terminal extremity. Following g-irradiation (IR), ATM-dependent phosphorylation of 53BP1 N-terminal extremity weakens the interaction of TIRR with 53BP1 and further promotes the recruitment of RIF1 on 53BP1 leading to the release of TIRR from 53BP1.
The importance of TIRR as a functional modulator of 53BP1 was clearly demonstrated in cells overexpressing TIRR. An excess of TIRR alters the stoichiometry and the dynamics of dissociation and consequently inhibits the recruitment of 53BP1 to DSBs. Physiological consequence of TIRR overexpression was observed in 2 scenarios, one it prevents class switching recombination in B lymphocytes - a process that largely relies on the recruitment of 53BP1 to endogenous DSBs generated during the recombination process. Second, it impairs PARP inhibitor sensitivity in BRCA1-mutated cells, mimicking an absence of 53BP1. Conversely, loss-of-function studies also reveal the impact of TIRR on 53BP1 activity. TIRR depletion destabilizes 53BP1 but more importantly affects how 53BP1 interacts with its partners. Indeed, the absence of TIRR increases the DNA-damage dependent association of 53BP1 with its effector partners such as RIF-1 or PTIP. This functionally correlates with the fact that depletion of TIRR renders BRCA1-mutated cells even more sensitive to PARP inhibitor in a 53BP1-dependent manner. This “hyper-active” state of 53BP1 has been also observed previously in cells overexpressing the E3 ubiquitin-ligase RNF168, an enzyme involved in the recruitment of 53BP1 to the damage.Citation6 Therefore, it is likely that TIRR protein level is tightly controlled in ‘normal’ cells to modulate 53BP1 function. Interestingly, a compilation of 50 studies in the Cancer Genome Atlas (TCGA) shows that the TIRR gene locus (alias Nudt16L1) is amplified in 29 out of the 34 different carcinomas. Therefore, one attractive possibility would be that amplification of the TIRR gene – that would result in TIRR protein level increase and consequently 53BP1 inactivation - is one of the mechanisms by which BRCA1-mutant tumors acquire PARP inhibitor resistance. Future analysis of BRCA1-mutant tumors from ovarian or breast cancer patients resistant to PARP inhibitors may reveal the clinical relevance of TIRR in cancer therapy.
The number of epigenetic reader proteins that are shown to be involved in disease development and progression is growing, and there is increasing knowledge about their involvement in the pathogenesis of various diseases. There is currently a significant effort in identifying small molecules that inhibit interactions between histone methyl lysine readers and their respective binding partners on chromatin. Initial inhibitors have emerged, but for many targets there are no known small-molecule ligands. TIRR is the first example of a bona fide cellular inhibitor of a histone methyl lysine reader and structural analyses suggest that the mechanism of masking the Tudor/methyl-lysine interface could be extrapolated to identify factors that inhibit the methyl-lysine binding function of other proteins. This is also a unique mechanism by which the activity of this class of proteins maybe broadly regulated. The molecular and structural details of the TIRR/53BP1 complex will be of great interest to the large community of researchers who work on other histone methyl lysine readers.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Zimmermann M, de Lange T. 53BP1: pro choice in DNA repair Trends Cell Biol 2014; 24:108-17; PMID:24094932; https://doi.org/10.1016/j.tcb.2013.09.003
- Chapman JR, Tayloy MR, Boulton SJ. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice Molecular Cell 2012; 47:495-510; PMID: 22920290; https://doi.org/10.1016/j.molcel.2012.07.029
- Drane P, Brault ME, Cui G, Meghani K, Chaubey S, Detappe A, Parnandi N, He Y, Zheng XF, Botuyan MV, Kalousi A, Yewdell WT, Munch C, Harper JW, Chaudhuri J, Soutoglou E, Mer G, Chowdhury D. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function Nature 2017; 543:211-6; PMID:28241136; https://doi.org/10.1038/nature21358
- Zhang A, Peng B, Huang P, Chen J, Gong Z. The p53-binding protein 1-Tudor-interacting repair regulator complex participates in the DNA damage response J Biol Chem 2017; 292, 6461-67; PMID:28213517; https://doi.org/10.1074/jbc.M117.777474
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair Cell 2006; 127:1361-73; PMID:17190600; https://doi.org/10.1016/j.cell.2006.10.043
- Zong D, Callen E, Pegoraro G, Lukas C, Lukas J, Nussenzweig A. Ectopic expression of RNF168 and 53BP1 increases mutagenic but not physiological non-homologous end joining Nucleic Acids Res 2015; 43:4950-61; PMID:25916843; https://doi.org/10.1093/nar/gkv336