
The serine and threonine kinase AKT (also known as protein kinase B, PKB) integrates inputs from growth factors and metabolic effectors to control key multifunctional signaling hubs via direct phosphorylation of substrates that control cell growth, proliferation and survival.Citation1 Among critical pro-survival mechanisms, autophagy is an evolutionarily conserved housekeeping pathway that functions to degrade and recycle macromolecules and organelles as part of basal cell metabolism as well as in response to specific stimuli.Citation2 Autophagic degradation is based on the activity of the lysosome, a membrane-enclosed organelle that contains a wide array of hydrolytic enzymes with different macromolecule specificities.
Our previous work has established that transcription factor EB (TFEB) acts as a master regulator of the autophagy-lysosome pathway (reviewed in Citationref. 3). Since the initial characterization of TFEB as a transcriptional modulator of the lysosomal system, cytoplasm-to-nucleus translocation of TFEB has been identified as the main mechanism underlying regulation of TFEB activity.Citation4 While several proof-of-concept studies have shown that exogenous expression of TFEB can counteract appearance and progression of pathological changes in animal models of diseases with a prominent intracellular storage component, clinical translation has been limited by the lack of suitable entry points to modulate TFEB activity that are amenable to pharmacological manipulation with available drugs.
Our recent work has uncovered AKT-mediated phosphorylation of TFEB as a mechanism by which AKT modulates the autophagy-lysosomal pathway.Citation5 AKT phosphorylates TFEB on serine 467, a residue that is evolutionarily conserved in vertebrate species and that is also conserved in TFEB's close homologs, TFE3 and MITF. A TFEB phosphomutant in which the AKT phosphoacceptor site was substituted by alanine to impede AKT-mediated phosphorylation (TFEB S467A) showed increased nuclear localization and stability as well as increased ability to activate TFEB downstream target genes, thus indicating that TFEB function is inhibited by AKT phosphorylation.Citation5 Accordingly, both AKT knockdown and pharmacological inhibition of AKT promoted TFEB nuclear translocation; AKT inhibition also activated TFE3 and MITF, thus revealing conservation of this regulatory pathway ().Citation5
Figure 1. AKT modulates the autophagy-lysosome pathway via TFEB. Phosphorylation of TFEB by AKT decreases nuclear TFEB. Inhibition of AKT activity using pharmacological inhibitors or trehalose promotes nuclear translocation of TFEB and activation of the autophagy-lysosome pathway and enhances the clearance of autophagic and lysosomal substrates.
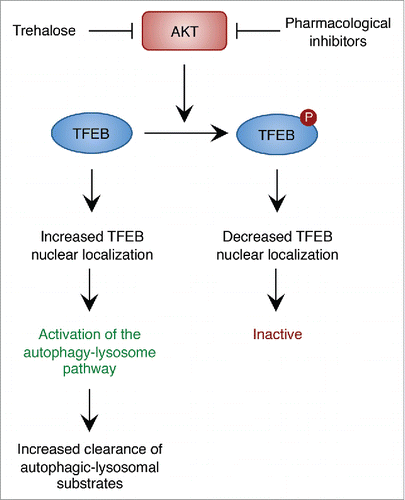
Notably, AKT inhibition resulted in increased number of LC3-positive autophagic vesicles and increased expression of autophagic and lysosomal genes, thereby indicating global enhancement of the autophagy-lysosome pathway. Similar results were obtained with trehalose, a natural disaccharide that stimulates autophagy and exerts neuroprotection in various models of neurodegenerative proteinopathies.Citation2 The mechanism of action of trehalose has been elusive for more than a decade; recent work has shown that trehalose inhibits the activity of glucose transporters at the plasma membrane, thus inducing a starvation-like state that promotes autophagy via the AMPK-ULK1 axis.Citation6 We have shown that trehalose inhibits AKT activity in vitro and in vivo, thus resulting in TFEB nuclear translocation and activation of the autophagy-lysosome pathway.Citation5 Importantly, expression of a constitutive active form of AKT prevented trehalose from inducing TFEB nuclear translocation, demonstrating that trehalose indeed activates TFEB via AKT inhibition.Citation5 Activation of the autophagy-lysosome pathway obtained by pharmacological inhibition of AKT or trehalose administration resulted in enhanced clearance of aberrant proteolipid aggregates in cells from patients with neuronal ceroid lipofuscinosis, a genetically diverse subgroup of the lysosomal storage diseases.Citation5 Significantly, oral administration of trehalose to a mouse model of juvenile neuronal ceroid lipofuscinosis (Batten disease) diminished AKT activity and promoted TFEB activity in the brain, again resulting in the activation of the autophagy-lysosome pathway and the subsequent clearance of ceroid lipopigments in neurons, which finally led to protection from neurodegeneration and elongation of the life span of affected mice.Citation5
These findings provide a direct, pharmacologically actionable entry point to modulate TFEB activity and enhance the autophagy-lysosome pathway through increased TFEB function. The AKT-TFEB signaling pathway might be leveraged to devise treatments of degenerative diseases in which there is pathological storage of aberrant aggregates of proteins or other macromolecules, such as in Alzheimer disease, Parkinson disease, Huntington's disease and lysosomal storage disorders. Clinical development of trehalose is currently focusing on the treatment of arterial aging (ClinicalTrial.gov ID: NCT01575288),Citation7 oculopharyngeal muscular dystrophy (NCT02147886), spinocerebellar ataxia 3 (NCT02147886) and bipolar depression (NCT02800161)—all conditions that could benefit from increased autophagy. Pharmacokinetic (PK) and pharmacodynamic (PD) profiles of trehalose need to be determined to move forward with the possible clinical application of trehalose in neurodegenerative diseases. On the other hand, pharmacological inhibition of AKT is undergoing clinical development for the treatment of various types of cancer using small drugs with significantly different PK/PD characteristicsCitation1 and might represent as well a possible route to activate the autophagy-lysosome pathway in degenerative storage disorders. Since AKT signaling is known to play key roles in neuronal growth, polarity and survival,Citation1 repositioning AKT pharmacological inhibitors for the long-term treatment of patients with chronic diseases such as neurodegenerative storage disorders will require the development of specific strategies to minimize toxicity and side effects. A possible strategy could focus on using comparatively low dosages of AKT inhibitors and patterns of sustained administration to enhance basal autophagy and improve cellular clearance up to therapeutic levels without compromising neuronal health. An alternative strategy could entail use of bursts of AKT inhibition followed by rest periods to counteract, over time, the slowly progressing accumulation of aberrant storage material to keep it to virtually non-pathogenic levels. Clearly, much preclinical work is necessary to determine the most viable route(s) to a clinical translation of AKT-mediated control of TFEB activity in human disease.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell 2017; 169:381-405; PMID:28431241; https://doi.org/10.1016/j.cell.2017.04.001
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; https://doi.org/10.1038/nm.3232
- Sardiello M. Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann N Y Acad Sci 2016; 1371:3-14; PMID:27299292; https://doi.org/10.1111/nyas.13131
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463; https://doi.org/10.1126/science.1174447
- Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, Chaudhury A, Bajaj L, Bondar VV, Bremner L, Saleem U, Tse DY, Sanagasetti D, Wu SM, Neilson JR, Pereira FA, Pautler RG, Rodney GG, Cooper JD, Sardiello M. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun 2017; 8:14338; PMID:28165011; https://doi.org/10.1038/ncomms14338
- DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, Chi M, Newberry EP, Chen Z, Finck BN, Davidson NO, Yarasheski KE, Hruz PW, Moley KH. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal 2016; 9:ra21; PMID:26905426; https://doi.org/10.1126/scisignal.aac5472
- Kaplon RE, Hill SD, Bispham NZ, Santos-Parker JR, Nowlan MJ, Snyder LL, Chonchol M, LaRocca TJ, McQueen MB, Seals DR. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging (Albany NY) 2016; 8:1167-83; PMID:27208415; https://doi.org/10.18632/aging.100962