
ABSTRACT
Organization of chromatin structure is indispensible to the maintenance of genome integrity. The retinoblastoma tumor suppressor protein (pRB) mediates both transcriptional repression and chromatin organization, but the independent contributions of these functions have been difficult to study. Here, we utilize a synthetic Rb1 mutant allele (F832A) that maintains pRB association at cell cycle gene promoters, but disrupts a cyclin-dependent kinase (CDK)-resistant interaction with E2F1 to reduce occupancy of pRB on intergenic chromatin. Reduced pRB chromatin association increases spontaneous γH2AX deposition and aneuploidy. Our data indicates that the CDK-resistant pRB-E2F1 scaffold recruits Condensin II to major satellite repeats to stabilize chromatin structure in interphase and mitosis through mechanisms that are distinct from silencing of repetitive sequence expression.
Introduction
Transitions between euchromatic and heterochromatic states accompany higher-order folding and compaction of chromatin to ensure proper coordination of transcription, DNA replication, and chromosome segregation. Changes to chromatin accessibility and compaction are achieved through enzymes that covalently modify DNA or histone tail residues, remodel nucleosomes, or mediate topological re-arrangement of chromosome architecture.Citation29 Changes to chromatin-organizing enzymes through somatic mutations, epigenetic silencing, or post-translational modifications disrupts coordinated chromatin accessibility and architecture. Misregulation of chromatin organization establishes a state of genome instability characterized by persistent DNA damage that often precedes tumorigenesis.Citation19 In this regard, it is not surprising that genes encoding broad organizers of chromatin structure are often classified as tumor suppressors or oncogenes.
In human cancer, a frequent target for disruption or misregulation is the retinoblastoma protein (pRB).Citation11 Primary fibroblasts deficient for pRB exhibit relaxed chromatin structure,Citation14 along with replication defects and reduced chromatin compaction in prophase and chromosome segregation errors.Citation9,10,21,24,25 These changes are often accompanied by manifestations of genome instability, such as aneuploidy and widespread DNA damage.Citation9,25,35 However, since pRB exerts transcriptional control over S- and M- phase cell cycle targets through E2Fs,Citation13,31 genetic models of pRB deficiency may not distinguish the mechanistic contributions of pRB chromatin regulation from pRB-E2F transcriptional control in the maintenance of genome integrity. Thus, the ability to determine contributions of pRB chromatin organization to maintenance of genome stability requires specific loss-of-function models that maintain E2F transcriptional control and G1-S cell cycle regulation.
To regulate chromatin structure, pRB serves as a scaffold for several regulatory enzymes that methylate DNA, modify histone tails, or mediate topological remodeling of nucleosomes.Citation20 DNA-binding proteins, such as transcription factors, mediate region-specific pRB-chromatin association.Citation33 This recruitment is predominantly mediated by E2F transcription factors. However, pRB-E2F complexes dissociate at the G1-S transition in response to cyclin-dependent kinase (CDK) phosphorylation. Recent evidence indicates that pRB uses an alternate CDK-resistant interaction with E2F1 to associate with repetitive elements.Citation5,7,9,16,17 Germline disruption of this interaction abrogates EZH2-mediated facultative heterochromatinization at repetitive elements in concert with repeat misexpression and lymphomagenesis in mice.Citation17 The cancer susceptibility observed upon loss of CDK-resistant pRB-chromatin association merits investigation into whether this pRB-E2F1 interaction might underlie other pRB-dependent activities that impact genome integrity and cancer susceptibility.
Here, we report that disruption of pRB's unique interaction with E2F1 causes the accumulation of γH2AX foci, aneuploidy, increased RPA phosphorylation, and mitotic defects. Importantly, E2F target genes involved in DNA replication and mitosis remain at normal expression levels. We demonstrate that cells bearing an F832A mutation in Rb1 (Rb1S) exhibit impaired chromatin recruitment of the Condensin II complex specifically at genomic locations where γH2AX accumulates. This mechanism further suggests that chromatin organization is an indispensible facet of pRB-mediated tumor suppression.
Results
The Rb1S mutation causes defects in genome integrity
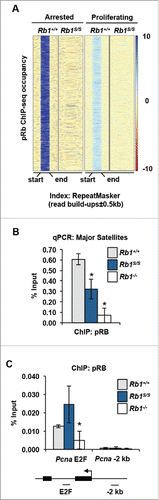
To assess the contribution of pRB-E2F1 interactions in chromatin association and its effects on genome stability, we used a described previously mutation (F832A, called Rb1S) that disrupts pRB's unique C-terminal interaction with the E2F1 marked box domain.Citation7,9,17 Importantly, this mutation preserves interactions between the pRB large pocket domain and the E2F transactivation domain to maintain proliferative control.Citation17 ChIP-sequence of pRB from wild type and Rb1S/S cells was analyzed. Alignment of pRB ChIP-seq reads reveals locations of pRB enrichment within repetitive elements throughout the genome and these are lost in ChIP-seq using pRBS (). Strikingly, this pattern of repeat occupancy by pRB is relatively preserved under proliferating conditions and can be detected at major satellite repeats using ChIP-qPCR (). Conversely, pRB ChIP-qPCR demonstrates that pRBS retains occupancy at cell cycle and E2F regulated promoters, such as Pcna, at levels comparable to that of wild type pRB ().Citation17 In addition, previous ChIP-qPCR and ChIP-seq analysis reveals similar results at other E2F responsive promoters.Citation17 Therefore, in contrast to previous models of pRB-deficiency, the Rb1S mutation permits assessment of post-G1 pRB-chromatin regulatory activities independent of E2F cell cycle gene association and regulation.
Figure 1. Disruption of pRB association with repetitive sequences. (A) Heat maps display pRB ChIP-seq read build ups at scaled wild type peak locations. pRB read build ups from wild type and Rb1S/S MEFs are shown for mm9 RepeatMasker index locations under arrested and proliferating conditions. Each row contains ± 0.5 kb of flanking sequence surrounding the scaled peak location. Intensity scales on right indicate the magnitude of read enrichment over input control. (B) ChIP-qPCR quantifies pRB enrichment at major satellites in proliferating MEFs of the indicated genotypes. (C) pRB ChIP-qPCR analyzing amplicons at the Pcna transcriptional start site (TSS) and 2 kb 5′ of Pcna. For all graphs, error bars indicate one standard deviation from the mean, and an asterisk represents a significant difference from wild type (n = 3, P ≤ 0.05 by t-test).
We investigated several broad measures of genome integrity in Rb1S/S MEFs relative to controls. Since Rb1−/− cells exhibit aneuploidy with increasing rounds of cell division,Citation32 we sought to determine whether loss of pRB-chromatin association at non-cell cycle genes could cause a similar effect. To assess cellular DNA content, proliferating and serum-starved MEFs were stained with propidium iodide and analyzed by flow cytometry. DNA content greater than the 4N peak was quantified to determine the proportion of aneuploid cells. In accordance with previous reports, Rb1−/− MEFs exhibit significantly elevated levels of > 4N DNA. Intriguingly, Rb1S/S MEFs also exhibit increased levels of > 4N DNA relative to wild type MEFs under arrested and proliferating growth conditions, albeit at levels less than those of Rb1−/− MEFs ().
Figure 2. Loss of genome integrity in Rb1S/(S)mutant cells. (A) MEFs stained with propidium iodide were analyzed by flow cytometry for DNA content. The histogram displays gates that demarcate populations with >4N DNA content that were quantified for the indicated MEF genotypes. Error bars indicate one standard deviation from the mean, and an asterisk represents a significant difference from wild type compared within the same growth condition (n = 3, P≤ 0.05 by t-test). (B) Immunofluorescence microscopy of proliferating MEFs stained for γH2AX (red) and DAPI (blue). White boxes indicate regions shown at increased magnification within the inset. Scale bars indicate relative magnification. (C) The number of γH2AX foci per nucleus was quantified and compared among 3 biological replicates per genotype using a χ2 test (Rb1+/+ n = 1054, Rb1S/S n = 744, Rb1−/− n = 602 total nuclei analyzed). (D) Western blots show RPA32 pSer33 levels for the indicated genotypes from arrested and proliferating MEFs. The Actin blot serves as a loading control. (E) RPA32 pSer33 and RPA32 western blots of chromatin fractions from proliferating MEFs. Histone H3 blot indicates loading.
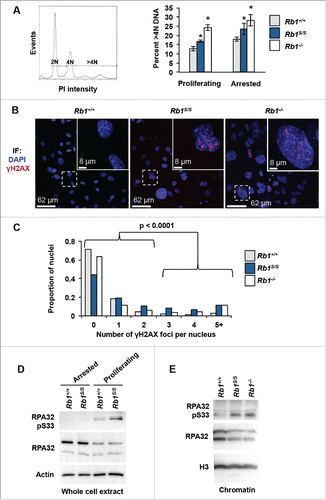
Another broad manifestation of genome instability observed upon pRB loss is the emergence of γH2AX foci in fluorescence microscopy experiments. Immunofluorescence revealed increased γH2AX foci in proliferating Rb1S/S MEF cultures comparable in magnitude to Rb1−/− MEFs, with approximately 10% of nuclei displaying 5 or more foci ( and ). Increased γH2AX foci together with elevated >4N DNA content in Rb1S/S cells suggests pRB-E2F1 complexes might mediate post-G1 functions. Post-G1, pRB reduces DNA damage through mitigation of replication stress via mechanisms that remain poorly understood.Citation2,9,25 To explore the effects of reduced pRB chromatin association on replication, we assessed relative phosphorylation levels of the single-strand DNA binding protein RPA. DNA damage response kinases such as ATR phosphorylate RPA32 on S33 to stabilize ssDNA, minimize damage and facilitate repair.Citation26 Western blots of whole cell extracts reveal increased levels of RPA32 pS33 under proliferating conditions relative to arrested wild type MEFs. This proliferation-dependent RPA32 pS33 increase appears further elevated in Rb1S/S MEFs relative to control cells (). Strikingly, western blots of chromatin fractions revealed that the elevated RPA32 pS33 in mutant MEFs can be detected on chromatin at levels comparable to that of Rb1−/− MEFs (). Increased RPA32 pS33 is indicative of a response to defects that arise during DNA replication in Rb1S/S MEFs.
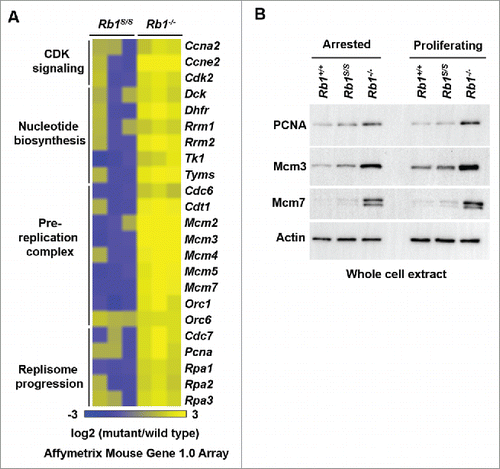
In pRB-deficient cells, misregulation of E2F transcriptional control of DNA replication components contributes to aneuploidy.Citation32 However, in accordance with pRBS association with cell cycle promoters in ChIP experiments (), microarray analysis generally revealed unchanged or diminished E2F targets involved in DNA replication in arrested Rb1S/S MEFs (). In contrast, Rb1−/− MEFs exhibited increased expression of DNA replication components under the same culture conditions (). Beyond mRNA levels, western blots of whole cell extracts confirmed expression levels of DNA replication components in Rb1S/S MEFs paralleled those in wild type MEFs under both arrested and proliferating conditions, and contrasted with the pronounced misregulation evident upon complete loss of pRB expression ().
Figure 3. Normal expression of DNA replication targets of pRB-E2F. (A) Expression microarrays were performed with RNA from serum-starved MEFs of the indicated genotypes and wild type controls (n = 3). For each gene listed, corresponding log2 values of each mutant replicate vs. wild type is shown as a heat map. (B) Western blots indicate PCNA, Mcm3, and Mcm7 levels detected in whole-cell extracts from arrested and proliferating MEFs. The Actin blot serves as a loading control.
Collectively, these observations suggest that reduced pRB-chromatin association under proliferating growth conditions increases the frequency of events that can compromise genome integrity. Expression analysis suggests that these effects in Rb1S/S MEFs are independent of E2F transcriptional control of DNA replication genes.
Rb1S mutant cells exhibit defects in mitosis
Beyond replication, pRB maintains proper chromosome segregation through coordination of multiple mitotic processes such as the regulation of E2F-activated spindle assembly checkpoint targets, and regulation of chromosome condensation and segregation.Citation23 We investigated whether the Rb1S mutation affected mitosis. To visualize mitotic progression, wild type, Rb1S/S, and Rb1−/− MEFs were transduced with GFP-tagged H2B using viral delivery, and subjected them to live-cell video microscopy. Analysis of Rb1S/S cells revealed a marked defect in chromosome organization that frequently preceded defective chromosome congression at metaphase ( and ). We next analyzed mitotic chromosome segregation. During anaphase, pRB-deficient cells exhibit elevated levels of chromosome bridges. Indeed, Rb1S/S, and Rb1−/− MEFs both exhibit significant increases in chromosome bridges relative to wild type cells ( and ). shows examples of normal chromosome segregation and cytokinesis observed by video microscopy in wild type MEFs. These microscopy experiments revealed numerous chromosome segregation defects present in Rb1S/S MEFs, such as partial segregation of chromosomes, missegregation of all chromosomes to one daughter before cytokinesis, or failure of cytokinesis leading to binucleated cells (). Ultimately, the majority of mitotic Rb1S/S and Rb1−/− MEFs fail to faithfully segregate duplicated chromosomes to their daughter cells, while over 90% of wild type MEFs displayed proper mitotic progression in this assay (). Curiously, chromosome missegregation describes the majority of segregation defects present in Rb1S/S MEFs, while Rb1−/− MEFs predominantly fail to undergo proper cytokinesis ().
Figure 4. Defective mitosis in Rb1S/(S)mutant cells. Mitosis was investigated by video microscopy using MEFs transduced with H2B-GFP (Rb1+/+ n = 19, Rb1S/S n = 15, Rb1−/− n = 11). Proportions of mitotic errors were compared using a χ2 test with significant differences indicated by an asterisk, while N.S. denotes comparisons that were not significantly different. Scale bars indicate relative magnification for all images. (A) Merged images of phase-contrast and GFP channels display examples of normal and defective chromosome congression in metaphase. (B) Quantitation of the frequency of congression errors observed per genotype. (C) Merged images of phase-contrast and GFP channels distinguish a normal anaphase cell from a cell that exhibits a chromosome bridge. (D) Quantitation of the frequency of chromosome bridges observed per genotype. (E) Merged images of phase-contrast and GFP channels to demonstrate examples of defective chromosome segregation and cytokinesis observed by video microscopy. (F) Quantitation of defective segregation events observed per genotype. (G) Fluorescence microscopy of proliferating MEFs stained with DAPI (blue). White arrows demark examples of micronuclei while scale bars indicate relative magnification. (I) Quantitation of micronuclei visualized by immunofluorescence microscopy. Error bars indicate one standard deviation from the mean, and an asterisk represents a significant difference from wild type (P ≤ 0.05 by t-test, n = 3).
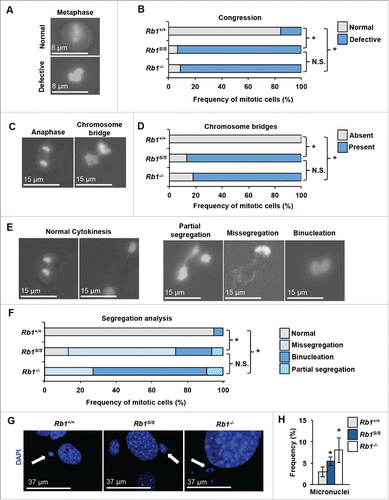
Mitotic errors such as chromosome bridges and missegregation events contribute to the generation of micronuclei.Citation15 To determine whether mitotic errors in Rb1S/S cells were associated with the accumulation of micronuclei, MEFs were fixed and stained with DAPI for fluorescence-based visualization. Quantification of micronuclei revealed a significant increase in Rb1S/S cells relative to wild type ( and ). This increase was marginally less than the magnitude of micronuclei present in Rb1−/− cells ().
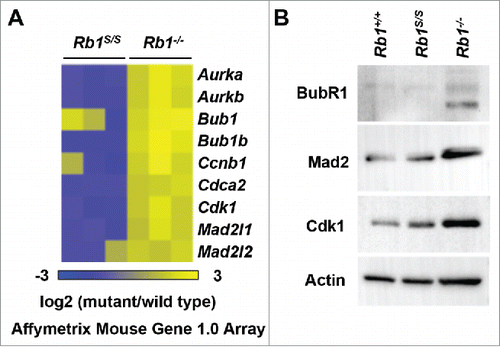
We next assessed whether misregulation of E2F mitotic target gene expression might underlie the mitotic errors observed upon reduction of pRB-chromatin association. Akin to DNA replication components, microarray analysis revealed normal expression of E2F mitotic checkpoint targets in Rb1S/S MEFs that contrasted with the misexpression observed in Rb1−/− MEFs (). Western blots of whole cell extracts confirmed that expression levels of mitotic checkpoint proteins in Rb1S/S MEFs resembled expression levels observed in wild type fibroblasts. Accordingly, Rb1−/− cells exhibit misexpression of E2F mitotic checkpoint targets at the protein level (). Overall, lack of mitotic checkpoint misexpression in the presence of mitotic errors suggests that pRB-chromatin association facilitates mitotic progression independently of E2F transcriptional control.
Figure 5. Proper regulation of mitotic pRB-E2F target genes. (A) Expression microarrays were performed with RNA from serum-starved MEFs of the indicated genotypes and wild type controls (n = 3). For each gene listed, corresponding log2 values of each mutant replicate vs. wild type is shown as a heat map. (B) Western blots indicate BubR1, Mad2, and Cdk1 levels detected in whole-cell extracts from proliferating MEFs. The Actin blot serves as a loading control.
pRB-E2F1 recruits Condensin II to major satellites to mitigate DNA damage
In light of the post-G1 defects apparent upon diminished pRBS chromatin association, we sought to identify a pRB-dependent mechanism that could link increased γH2AX, increased RPA32 pS33, and mitotic defects observed in Rb1S/S cells. Since pRB serves as a recruitment factor for numerous chromatin regulators including Condensin II,Citation9,10,21 we investigated its recruitment to sites of γH2AX deposition. First, to determine whether Condensin II exhibited sensitivity to diminished pRB-chromatin association, we assessed the expression of Condensin II subunits. Western blots of whole cell extracts from arrested and proliferating MEFs revealed no differences in CAP-D3 and SMC2 expression in mutant cells relative to wild type controls (). We next assessed whether the Rb1S mutation affected Condensin II recruitment to chromatin. Western blots of chromatin fractions revealed that relative to the arrested state, both CAP-D3 and SMC2 exhibited increased chromatin loading in proliferating wild type MEFs. Notably, this proliferation-dependent increase in Condensin II chromatin loading is diminished in Rb1S/S cells (). We then explored whether reduced Condensin II loading on chromatin in mutant cells was due to a direct pRB-mediated recruitment mechanism. ChIP for CAPD3 at pericentric major satellite repeats reveals reduced occupancy in Rb1S/S MEFs relative to wild type levels (). ChIP for CAP-D3 followed by ChIP for pRB confirms that CAP-D3 localizes with pRB to pericentric major satellite repeats in wild type MEFs (). The reciprocal ChIP-reChIP experiment recapitulates this trend (). Finally ChIP-reChIP reveals significantly reduced CAP-H2-pRB co-recruitment to major satellites in Rb1S/S MEFs, consistent with the global reduction of Condensin II loading observed in chromatin fractions ().
Figure 6. Recruitment of CAP-D3 by pRB mitigates DNA damage at pericentromeric major satellite repeats. (A) Western blots of whole-cell extracts from arrested and proliferating MEFs were used to determine CAP-D3 and SMC2 levels. The Actin blot serves as a loading control. (B) CAP-D3 and SMC2 western blots of chromatin fractions. Coomassie-stained histones serve as a loading control. (C) CAP-D3 ChIP-qPCR performed using chromatin from proliferating MEFs and quantified at major satellite repeats. (D) CAP-D3-pRB ChIP-reChIP performed using chromatin from proliferating MEFs was quantified at major satellite repeats by qPCR. (E) pRB-CAP-D3 ChIP-reChIP performed as described for , but with a reciprocal order of ChIPs. (F) CAP-H2-pRB ChIP-reChIP performed as described for . (G) γH2AX ChIP-qPCR was performed using chromatin from proliferating MEFs and quantified at pericentromeric major satellite repeats and normalized to IgG ChIP performed in parallel from the same chromatin preparation. ((H)and I) γH2AX ChIP-qPCR from quantified at L1 5′ UTRs and IAP LTRs. For all graphs, error bars indicate one standard deviation from the mean, and an asterisk represents a significant difference from wild type (n = 3, P ≤ 0.05 by t-test).
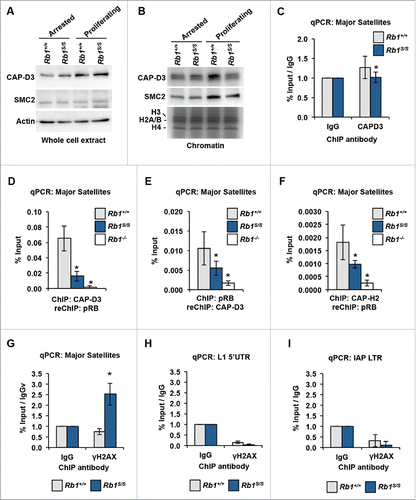
Condensin II-mediated regulation of replication and chromosome condensation requires chromatin loading. We hypothesized that genomic locations of pRB-dependent Condensin II association, such as pericentric repeats, would be particularly sensitive to accumulation of γH2AX upon reduction of Condensin II recruitment. ChIP-qPCR demonstrates a relative increase in γH2AX at major satellite repeats in proliferating Rb1S/S MEFs, while γH2AX levels in wild type MEFs do not exceed background levels (). By comparison, γH2AX ChIP-qPCR experiments demonstrated that DNA damage is not increased at LINE1 or endogenous retroviral repeats in Rb1S/S cells ( and ). This indicates that only specific genomic locations accumulate γH2AX in response to the Rb1S mutation rather than all repeat locations occupied and regulated by pRB. Overall, site-specific γH2AX accumulation at locations of pRB-Condensin II loss in Rb1S/S MEFs suggests that the CDK-resistant pRB-E2F1 complex mediates Condensin II recruitment to pericentromeric repeats to support genome integrity. Loss of this recruitment event is associated with the onset of DNA damage and mitotic errors that are prevalent in Rb1S/S MEFs, and the contributions of this mechanism to cancer susceptibility in Rb1S/S mice is likely significant.
Discussion
The original null alleles of Rb1 in mice revealed that it was indispensable to the maintenance of genome integrity.Citation38 However, deregulation of E2F cell cycle targets concurrent with loss of genome stability hindered the ability to assess whether pRB could maintain genome integrity independent of E2F transcriptional control. Here, we used a synthetic Rb1 mutant allele that maintains pRB association at E2F cell cycle targets, but reduces overall pRB-chromatin association at intergenic regions.Citation17 We demonstrate that cells from these mice possess inherently unstable genomes characterized by markers of DNA damage, replication abnormalities, and aneuploidy.
Elevated RPA32 pS33 in Rb1S/S chromatin is suggestive of DNA replication defects. pRB-E2F1-dependent Condensin II recruitment prevents many features that are characteristic of ‘under replication’ of pericentric repeats.Citation9,12,22 It remains to be explored whether the pRB-E2F1 scaffold underlies other pRB interactions with replication components that may also contribute to spontaneous DNA damage. pRB restricts DNA replication initiation and progression through its interaction with Mcm7 and its ability to suppress DNA polymerase α and Ctf4 recruitment to replisomes.Citation3 It has also been shown that pRB associates with Orc1 at replication origins.Citation1,27 Finally, pRB can displace PCNA to halt progression of the replication fork.Citation4 If the pRB-E2F1 scaffold mediates these interactions, it is possible that they also contribute to RPA32 pS33 and γH2AX at pericentric repeats.
We note a significant failure of Rb1S/S cells to condense chromosomes that precedes numerous chromosome segregation defects. These defects parallel mitotic errors observed in other models that are deficient for pRB-Condensin II recruitment to chromatin.Citation10,21,24 Collectively, these data suggest that pRB-E2F1 forms a scaffold to permit Condensin II interaction with the pRB LxCxE binding cleft. Despite characterization of chromatin recruitment, mechanistic details of pRB-Condensin II association remain poorly understood. However, these phenotypes and the ensuing lymphoma in Rb1S/S mice has intriguing similarities to recent studies examining a hypomorphic CAP-H2 mutant mouse strain (called Nessy),Citation36 as well as lymphomas from E2f1−/− mice.Citation37 E2f1−/− mice frequently succumb to follicular B-lymphomas evident in the mesenteric lymph node and this is a common finding in Rb1S/S mice.Citation17,37 Nessy, or Caph2I15N mutant mice, have lineage-specific delayed anaphase entry that manifests as aneuploid CD4+/lo;CD8+;CD71+ thymic T-cells that develop into lymphomas in adult animals.Citation36 It will be interesting to understand the subtle differences in tissue tropism between the Rb1S/S and Nessy mice, although other pRB-E2F1 containing complexes recruited to chromatin likely underlie some of these differences.
Our observations contribute to a growing body of literature that challenges classification of pRB as ‘inactive’ beyond entry into S-phase. In addition to Condensin II, the pRB-E2F1 scaffold also recruits EZH2 to repetitive elements in cycling cells.Citation17 We envision that disruption of both complexes likely contributes to the cancer phenotype in Rb1S/S mice. First, loss of Condensin II recruitment and instability phenotypes are found in Rb1 mutant mice with defective LxCxE interactions (Rb1L), but this defect is insufficient to trigger cancer on its own.Citation9 In this study we show that instability phenotypes found in Rb1S/S cells are most attributable to defects in Condensin II recruitment as γH2AX accumulates preferentially in pRB-E2F1-Condensin II locations, yet Rb1S/S mice are cancer prone. An important difference between Rb1S/S and Rb1L/L mutant mice is repeat misexpression that predominantly occurs in the spleens of Rb1S/S mice where the majority of tumors arise. Based on this reasoning, we expect that loss of both pRB-E2F1-Condensin II and pRB-E2F1-EZH2 complexes caused by the F832A mutation are both necessary to cause the cancer phenotype observed in Rb1S/S mice. Collectively, this body of work further emphasizes the multifunctionality of pRB and how its disparate activities work together to underlie its tumor suppressor role.
Experimental procedures
Cell culture
Gene targeted mouse strains bearing a null allele of Rb1 (Rb1tm1Tyj) and an F832A point mutation to disrupt pRB's unique interaction with E2F1 (Rb1S, or Rb1tm3Fad) have been described previously.Citation17,18 Primary murine embryonic fibroblasts (MEFs) were isolated and cultured from E13.5 embryos of the desired genotypes according to established procedures.Citation34 All experiments were performed on passage 4 (P4) MEFs from at least 3 different embryo isolates.
Fluorescence microscopy
Immunofluorescence was conducted as described previously.Citation9 Briefly, 2.5 × 105 MEFs were seeded per confocal dish, incubated 24h, washed 3x for 5 minutes in 1x PBS, fixed by incubation with 4% paraformaldehyde in 1x PBS for 10 minutes at room temperature, then washed and stored at 4°C. To immunostain, cells were permeabilized for 10 minutes with 0.3% Triton X-100 in 1xPBS (PBS-T), blocked for 1 hour at room temperature with 5% goat serum in PBS-T, incubated one hour with mouse anti-Phospho histone H2A.X Ser139 (05–636, Millipore) diluted 1:400 in PBS-T, washed, and incubated one hour in goat anti-mouse Alexa Fluor 594 secondary antibody diluted 1:800 in PBS-T. Cells were washed 3x for 5 minutes with PBS-T, then incubated 5 minutes with DAPI in PBS-T. Stained MEFs were washed 3x with PBS-T, twice with 1x PBS, then mounted with a coverslip using SlowFade® Gold antifade reagent (S36937, Life Technologies). Fluorescence was visualized using an Olympus Fluoview FV1000 confocal microscope system, with images compiled using Olympus Fluoview FV1000 Viewer. γH2AX foci were quantified using the focinator programCitation28 with fixed thresholds and parameters maintained across all groups in a given experiment.
Flow cytometry
Asynchronous or serum starved MEFs were ethanol-fixed, stained with propidium iodide, treated with RNase, strained, and analyzed by flow cytometry on a Beckman-Coulter EPICS XL-MCL.Citation6
Detection of RNA and protein levels
RNA levels were determined by Affymetrix Mouse Gene 1.0 Array as described previously using total RNA from serum starved P4 MEFs.Citation8,17 Log2 values of mutant/wild type expression determined with Partek Genomics Suite were plotted as heat maps using matrix to PNG at chibi.ubc.ca/matrix. Array CEL files are available at GSE85640 and GSE54924. To assess protein levels, whole-cell lysates and chromatin fractions were generated as described previously,Citation17 resolved by SDS-PAGE, and immunoblotted using the following antibodies: RPA32 (A300–244A, Bethyl laboratories), RPA32 pSer33 (A300–246A, Bethyl laboratories), H3, (ab1791, Abcam), Actin (A2066, Sigma), PCNA (F-2 Santa Cruz), Mcm3 (4012S, Cell Signaling), Mcm7 (H-5, Santa Cruz), BubR1 (C-20, Santa Cruz), Cdk1 (ab7953 abcam), Mad2 (C-19, Santa Cruz), CAP-D3,Citation10 SMC2.Citation9
Chromatin immunoprecipitation and ChIP-seq analysis
Chromatin immunoprecipitation experiments and analyses were conducted as described previously.Citation17 Briefly, cross-linked chromatin fragments were pre-cleared with protein G Dynabeads for immunoprecipitation with ChIP antibodies. Cross-links were reversed at 65°C followed by DNA isolation. For ChIP-reChIP experiments, crosslinks were maintained following the first immunoprecipitation. Protein-DNA complexes were eluted, and subjected to a second immunoprecipitation with a different antibody. DNA was isolated as described for single-IP ChIP. deepTools was used to generate heat maps depicting ChIP-seq read enrichment at wild type peak locations. bamCompare was used to normalize ChIP to input reads, then computeMatrix was used to determine read enrichment at wild type repeat peak intersects with the mm9 RepeatMasker index from UCSC Table Browser. heatMapper was used to plot enrichment per peak location.Citation30 Reads are available at GSE85640. ChIP antibodies used were anti-phospho histone H2A.X Ser139 (07–164, Millipore), anti-CAP-D3,Citation10 anti-CAP-H2 (A302–275A, Bethyl laboratories) and a described previously cocktail of pRB antibodies.Citation8 ChIP-qPCR Primer sources have been described previously.Citation8,17 Sequences 5- to 3′ are as follows: PCNA_E2F_F CAGAGTAAGCTGTACCAAGGAGAC, PCNA_E2F_R CGTTCCTCTTAGAGTAGCTCTCATC, PCNA_-2kb_F CATCAGTGAATACGTCTCTGTTCCA, PCNA_-2kb_R CTGCTTCTCAGTTGTTTTAGGAAGG, Maj_F GACGACTTGAAAAATGACGAAATC, Maj_R CATATTCCAGGTCCTTCAGTGTGC, L1 5′ UTR_F CTGCCTTGCAAGAAGAGAGC, L1 5′ UTR_R AGTGCTGCGTTCTGATGATG, IAP LTR_F CTGACAGCTGTGTTCTAAGTGGTAAACAAA, IAP LTR_R AGAACACCACAGACCAGAATCTTCTGC
Video microscopy
Video microscopy was conducted as described previously.Citation10 Briefly, MEFs infected with ecotropic retroviruses carrying pBABE-H2B-GFP packaged in Bosc23 cells were plated into glass-bottom tissue culture dishes in phenol-free DMEM with 5% FBS, penicillin, streptomycin, and glutamine. Cells were maintained at 37°C with 5% CO2 as phase-contrast and fluorescent images were captured every 3 minutes for 15 hours by a DMI 6000b Leica microscope. Images were assembled into movies using Velocity, and analyzed for mitotic errors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank colleagues in the London Regional Cancer Program for frequent stimulating discussions during the course of this work.
Funding
CaRTT supported CAI and CHC. CHC was supported by a CIHR doctoral scholarship. CAI was the recipient of a WORLDiscoveries scholarship. FAD is the Wolfe Senior Fellow in Tumor Suppressor Genes at Western University. This work was supported by CIHR grants (MOP-89765 and MOP-64253) held by FAD.
References
- Avni D, Yang H, Martelli F, Hofmann F, ElShamy WM, Ganesan S, Scully R, Livingston DM. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol Cell 2003; 12:735-46; PMID:14527418; https://doi.org/10.1016/S1097-2765(03)00355-1
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011; 145:435-46; PMID:21529715; https://doi.org/10.1016/j.cell.2011.03.044
- Borysov SI, Nepon-Sixt BS, Alexandrow MG. The N terminus of the retinoblastoma protein inhibits DNA replication via a bipartite mechanism disrupted in partially penetrant retinoblastomas. Mol Cell Biol 2015; 36:832-45; PMID:26711265; https://doi.org/10.1128/MCB.00636-15
- Braden WA, Lenihan JM, Lan Z, Luce KS, Zagorski W, Bosco E, Reed MF, Cook JG, Knudsen ES. Distinct action of the retinoblastoma pathway on the DNA replication machinery defines specific roles for cyclin-dependent kinase complexes in prereplication complex assembly and S-phase progression. Mol Cell Biol 2006; 26:7667-81; PMID:16908528; https://doi.org/10.1128/MCB.00045-06
- Calbo J, Parreno M, Sotillo E, Yong T, Mazo A, Garriga J, Grana X. G1 cyclin/cyclin-dependent kinase-coordinated phosphorylation of endogenous pocket proteins differentially regulates their interactions with E2F4 and E2F1 and gene expression. J Biol Chem 2002; 277:50263-74; PMID:12401786; https://doi.org/10.1074/jbc.M209181200
- Cecchini MJ, Amiri M, Dick FA. Analysis of cell cycle position in mammalian cells. J Vis Exp 2012; 59:e3491
- Cecchini MJ, Dick FA. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem J 2011; 434:297-308; PMID:21143199; https://doi.org/10.1042/BJ20101210
- Cecchini MJ, Thwaites M, Talluri S, Macdonald JI, Passos DT, Chong JL, Cantalupo P, Stafford P, Saenz-Robles MT, Francis SM, et al. A retinoblastoma allele that is mutated at its common E2F interaction site inhibits cell proliferation in gene targeted mice. Mol Cell Biol 2014; 34:2029-45; PMID:24662053; https://doi.org/10.1128/MCB.01589-13
- Coschi C, Ishak C, Gallo D, Marshall A, Talluri S, Wang J, Cecchini M, Martens A, Percy V, Welch I, et al. Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov 2014; 4:840-53; PMID:24740996; https://doi.org/10.1158/2159-8290.CD-14-0215
- Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, Dick FA. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev 2010; 24:1351-63; PMID:20551166; https://doi.org/10.1101/gad.1917610
- Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev 2016; 30:1492-502; PMID:27401552; https://doi.org/10.1101/gad.282145.116
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 2011; 193:97-108; PMID:21444690; https://doi.org/10.1083/jcb.201011083
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 2004; 430:797-802; PMID:15306814; https://doi.org/10.1038/nature02820
- Herrera RE, Chen F, Weinberg RA. Increased histone H1 phosphorylation and relaxed chromatin structure in Rb-deficient fibroblasts. Proc Natl Acad Sci USA 1996; 93:11510-5; https://doi.org/10.1073/pnas.93.21.11510
- Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep 2012; 13:501-14; PMID:22565320; https://doi.org/10.1038/embor.2012.55
- Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, Haigis K, Gulino A, Lees JA. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell 2009; 15:184-94; PMID:19249677; https://doi.org/10.1016/j.ccr.2009.01.026
- Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, Ferwati S, MacDonald WA, Howlett CJ, Welch ID, et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol Cell 2016; 64:1074-87; PMID:27889452; https://doi.org/10.1016/j.molcel.2016.10.021
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature 1992; 359:295-300; PMID:1406933; https://doi.org/10.1038/359295a0
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; https://doi.org/10.1038/nature08467
- Longworth MS, Dyson NJ. pRb, a local chromatin organizer with global possibilities. Chromosoma 2010; 119:1-11; PMID:19714354; https://doi.org/10.1007/s00412-009-0238-0
- Longworth MS, Herr A, Ji JY, Dyson NJ. RBF1 promotes chromatin condensation through a conserved interaction with the Condensin II protein dCAP-D3. Genes Dev 2008; 22:1011-24; PMID:18367646; https://doi.org/10.1101/gad.1631508
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 2011; 13:243-53; PMID:21317883; https://doi.org/10.1038/ncb2201
- Manning AL, Dyson NJ. RB: mitotic implications of a tumour suppressor. Nat Rev Cancer 2012; 12:220-6; PMID:22318235
- Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev 2010; 24:1364-76; https://doi.org/10.1101/gad.1917310
- Manning AL, Yazinski SA, Nicolay B, Bryll A, Zou L, Dyson NJ. Suppression of genome instability in pRB-Deficient cells by enhancement of chromosome cohesion. Mol Cell 2014; 53:993-1004; PMID:24613344; https://doi.org/10.1016/j.molcel.2014.01.032
- Marechal A, Zou L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res 2015; 25:9-23; PMID:25403473; https://doi.org/10.1038/cr.2014.147
- Mendoza-Maldonado R, Paolinelli R, Galbiati L, Giadrossi S, Giacca M. Interaction of the retinoblastoma protein with Orc1 and its recruitment to human origins of DNA replication. PLoS One 2010; 5:e13720; PMID:21085491; https://doi.org/10.1371/journal.pone.0013720
- Oeck S, Malewicz NM, Hurst S, Rudner J, Jendrossek V. The Focinator - a new open-source tool for high-throughput foci evaluation of DNA damage. Radiation Oncol (London, England) 2015; 10:163; PMID:26238507; https://doi.org/10.1186/s13014-015-0453-1
- Papamichos-Chronakis M, Peterson CL. Chromatin and the genome integrity network. Nat Rev Genet 2013; 14:62-75; PMID:23247436; https://doi.org/10.1038/nrg3345
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 2014; 42:W187-191; PMID:24799436; https://doi.org/10.1093/nar/gku365
- Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011; 19:701-14; PMID:21665145; https://doi.org/10.1016/j.ccr.2011.04.017
- Srinivasan SV, Mayhew CN, Schwemberger S, Zagorski W, Knudsen ES. RB loss promotes aberrant ploidy by deregulating levels and activity of DNA replication factors. J Biol Chem 2007; 282:23867-77; PMID:17556357; https://doi.org/10.1074/jbc.M700542200
- Talluri S, Dick FA. Regulation of transcription and chromatin structure by pRB: here, there and everywhere. Cell Cycle 2012; 11:3189-98; PMID:22895179; https://doi.org/10.4161/cc.21263
- Thwaites MJ, Coschi CH, Isaac CE, Dick FA. Cell synchronization of mouse embryonic fibroblasts. Methods Mol Biol 2016; 1342:91-9; PMID:26254919
- van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, Joenje H, te Riele H. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev 2010; 24:1377-88; https://doi.org/10.1101/gad.580710
- Woodward J, Taylor GC, Soares DC, Boyle S, Sie D, Read D, Chathoth K, Vukovic M, Tarrats N, Jamieson D, et al. Condensin II mutation causes T-cell lymphoma through tissue-specific genome instability. Genes Dev 2016; 30:2173-86; PMID:27737961; https://doi.org/10.1101/gad.284562.116
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson N. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 1996; 85:537-48; PMID:8653789; https://doi.org/10.1016/S0092-8674(00)81254-4
- Zheng L, Flesken-Nikitin A, Chen PL, Lee WH. Deficiency of Retinoblastoma gene in mouse embryonic stem cells leads to genetic instability. Cancer Res 2002; 62:2498-502; PMID:11980640