
ABSTRACT
Triple negative breast cancer (TNBC) is a highly aggressive breast cancer subtype that lacks effective targeted therapies. Although TNBC is not defined by specific therapeutic targets, a subset of patients have tumors that overexpress cyclins. High cyclin D/E expression catalyzes CDK4/2 activity. In turn, CDK4/2 can non-canonically phosphorylate Smad3, a key TGFβ signaling intermediate, and this phosphorylation has been associated with the shift from tumor-suppressive to oncogenic TGFβ pathway action in breast oncogenesis. Additionally, CDK-mediated Smad3 phosphorylation facilitates an interaction between Smad3 and Pin1, a cis-trans isomerase that is also overexpressed in aggressive breast cancers. Treatment with CYC065, a CDK2/9 inhibitor, decreased non-canonical Smad3 phosphorylation and inhibited the Pin1-Smad3 interaction. We hypothesized that the interaction of Pin1 and Smad3, facilitated by CDK-mediated Smad3 phosphorylation, promotes TNBC cell aggressiveness. Inhibition of the Pin1-Smad3 interaction in TNBC cell lines, through depletion of Pin1 or CYC065 treatment, resulted in decreased cell migration/invasion and impeded the EMT program. Inhibition of CDK-mediated phosphorylation of Smad3 by mutagenesis also decreased cell migration, underscoring the importance of non-canonical CDK2 phosphorylation of Smad3 to enable cell motility. Pin1 depletion restored Smad3 protein levels and tumor-suppressive activity, suggesting that the Pin1-Smad3 interaction has a negative impact on canonical Smad3 action. Collectively, the data show that the Pin1-Smad3 interaction, facilitated by CDK-mediated Smad3 phosphorylation, is associated with oncogenic TGFβ signaling and breast cancer progression. Inhibition of this interaction with CYC065 treatment may provide an important therapeutic option for TNBC patients.
Introduction
Triple negative breast cancer (TNBC) is an aggressive breast cancer subtype. Characterized by the absence of hormone receptor and Her2 receptor expression, it is unresponsive to anti-hormonal and Her2-targeted therapies. Currently, there are no tailored therapies available for triple negative disease. Consequently, TNBC patients undergo surgery, chemotherapy, and radiation with limited success and are prone to relapse and death because of the tendency of TNBCs to metastasize.Citation1,2
Although TNBC is highly heterogeneous, a subset of patients with this disease overexpresses cyclins. Cyclin overexpression correlates with tumor stage/grade, increased aggressiveness and recurrence, and decreased survival time.Citation3-7 High cyclin expression catalyzes CDK activity, which can accelerate cell cycle progression. The prevalence of cyclin overexpression in cancer has fostered research into therapeutic CDK inhibitors, though the mechanism of action of these agents is complex.Citation8-11 Accordingly, CDK2 and CDK4 knockout mice are viable, indicating that these CDKs are not irreplaceable for cell cycle progression, and more recently these CDKs were identified as having a non-canonical role in TGFβ-mediated carcinogenesis.Citation12,13 CDK4/2 can phosphorylate Smad3, a critical intracellular mediator of the TGFβ pathway.Citation14,15 TGFβ signaling has a proposed dichotomous role in breast cancer; it can promote tumor suppression in well-differentiated cells, but switches to support tumor growth and metastasis in poorly differentiated cells. CDK4/2 phosphorylates Smad3 at sites Thr8, Thr179 and Ser213 in vivo, as well as Ser203 and Ser207 in vitro.Citation15 Research in our laboratory has shown that inhibiting phosphorylation of these sites rescues Smad3 tumor-suppressive activity and inhibits TNBC cell migration, highlighting the critical role of linker region phosphorylation in mediating the change from tumor-suppressive to oncogenic TGFβ signaling.Citation16-18 CDK2 and 4 inhibitors have been shown to prevent non-canonical Smad3 phosphorylation and to restore tumor-suppressive signaling, with CDK2 inhibition being highly effective in TNBC cells. Moreover, in a murine model, CDK2 inhibitor (CKD2i) treatment reduced TNBC tumor volume in a manner comparable to current chemotherapeutics, underscoring their potential for clinical use.Citation18
Phosphorylation at the Thr79 site (T179) of Smad3 is associated with Smad3 ubiquitination and degradation, which may contribute to enhanced cancer cell proliferation.Citation19–21 Moreover, pT179 is associated with Pin1-Smad3 binding and consequential promotion of migration and invasion in prostate cancer cells.Citation20,22 Pin1 is a peptidyl-prolyl cis/trans isomerase that recognizes the phosphorylated serine/threonine-proline motifs of its targets, including Smad3. Pin1 is overexpressed in breast and other cancers, and its expression correlates with increased breast tumor grade and poor patient outcomes.Citation23-25
From these findings, we hypothesized that the interaction of Pin1 and Smad3, facilitated by CDK-mediated Smad3 phosphorylation, promotes cancer cell growth, migration, and invasion in cyclin-expressing TNBC. In this report, we show that inhibiting CDK2-mediated phosphorylation of Smad3 blocks the Pin1-Smad3 interaction and reduces TNBC cell migration and invasion through changes in EMT-related signaling factors. Additionally, the use of CYC065 (Cyclacel), a CDK2/9 inhibitor in clinical development,Citation26 is able to inhibit TNBC cell migration/invasion. We further show that Pin1 has a negative impact on Smad3 protein stability and canonical tumor-suppressive Smad3 activity. Our study reveals a novel mechanism of oncogenic Smad3/TGFβ signaling and proposes a potential therapeutic to oppose this mechanism.
Results
Knock-down of Pin1 decreases TNBC cell migration and invasion
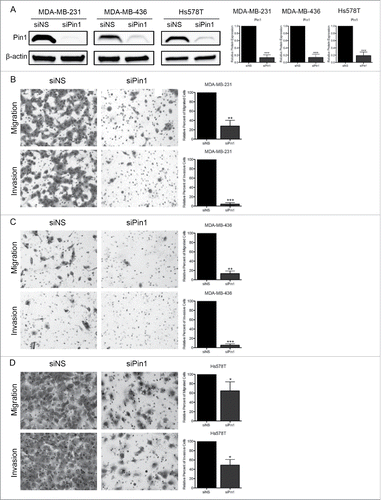
We explored the role of Pin1 in relation to Smad3 in the migratory and invasive ability of TNBC cells. To inhibit Pin1, Pin1 expression was knocked-down (KD) with Pin1-specific siRNA (siPin1) in 3 distinct TNBC cell lines, MDA-MB-231, MDA-MB-436, and Hs578T cells. These cells express cyclin D/E and CDK4/2.Citation18 Cells were transfected with non-specific siRNA (siNS) as a control. Pin1 expression was consistently depleted by greater than 80% in all cell lines by 96 hours post-transduction of siPin1 (). Thus, all Pin1 KD experiments were conducted 96 hours post-KD. Following Pin1 depletion, we assessed cell migration through transwell membranes and invasion through Matrigel-coated transwell membranes. Although each cell line was noted to have a different motility potential, with MDA-MB-436 cells being the least motile and Hs578T cells being the most motile, we found that depleting Pin1 expression resulted in a significant decrease in cell migration and invasion in all 3 cell lines (, , ).
Figure 1. KD of Pin1 expression significantly reduces TNBC cell migration and invasion. (A) Immunoblots of Pin1 96 hours-post KD demonstrating successful Pin1 depletion. Migration and invasion assays were performed 96 hours post-Pin1 KD with (B) MDA-MB-231, (C) MDA-MB-436, and (D) Hs578T cells. Representative images were taken at 100X total magnification, and the relative number of migrated/invaded cells from at least 3 independent experiments is graphed. *p < 0.05, **p < 0.01, ***p < 0.001.
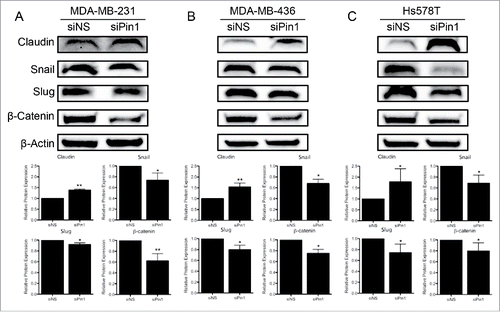
The decrease in cell motility corresponded to changes in EMT-associated protein levels. Expression of the tight-junction protein Claudin increased significantly with Pin1 KD, and expression of Snail and Slug significantly decreased. Expression of β-catenin, a key player in the WNT pathway, also decreased significantly following Pin1 depletion (). Thus, we conclude that Pin1 has a role in the ability of TNBC cells to promote EMT and migrate/invade.
Figure 2. KD of Pin1 expression in TNBC cells hinders the EMT program. Expression of the EMT-associated proteins Claudin, Snail, Slug, and β-catenin were quantified by immunoblot 96 hours post-Pin1 KD in (A) MDA-MB-231, (B) MDA-MB-436, and (C) Hs578T cells. Representative immunoblots are shown with densitometry analysis of at least 3 independent experiments. *p < 0.05, **p < 0.01.
Inhibition of CDK-mediated Smad3 phosphorylation inhibits Pin1-Smad3 binding and reduces TNBC migration/invasion
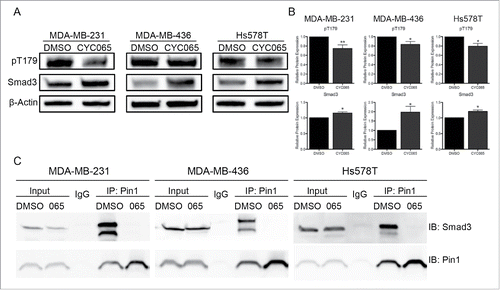
We next examined the mechanism by which Pin1 impacts TNBC cell migration and invasion. We hypothesized that the interaction of Pin1 with Smad3, dependent on CDK2 phosphorylation of Smad3, helps to facilitate cell migration and invasion. To inhibit CDK2-dependent Smad3 phosphorylation, we treated the 3 TNBC study cell lines with CYC065, a CDK2/9 inhibitor currently in clinical development. Treatment with CYC065 significantly reduced phosphorylation at T179 when compared with control DMSO treatment. Also, total Smad3 levels increased with CYC065 treatment ( and ). We subjected CYC065-treated cells to an endogenous co-immunoprecipitation assay. Lysate was incubated with Pin1 antibody-conjugated resin, and the elutant was analyzed by immunoblot for total Smad3. With DMSO control treatment, we saw a strong Pin1-Smad3 interaction, and treatment with CYC065 eliminated any discernable interaction (). There was no change in total Pin1 expression with CYC065 treatment ().
Figure 3. Inhibiting CDK-mediated Smad3 phosphorylation in TNBC cells blocks Pin1-Smad3 binding. (A) Representative immunoblots of pT179 and total Smad3 48 hours post-CYC065 treatment. (B) Densitometry analysis of immunoblots of at least 3 independent experiments. *p < 0.05, **p < 0.01. (C) Co-immunoprecipitation of Smad3 with Pin1 following CYC065 treatment. Pin1 antibody-conjugated resin was used to isolate Pin1 and interaction was analyzed by immunoblot for Smad3.
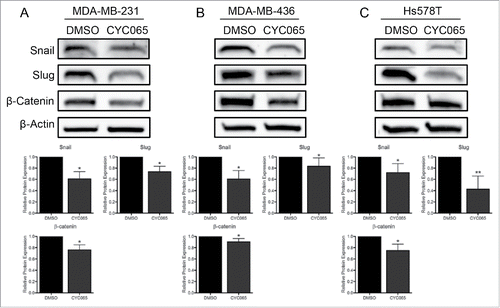
To determine if inhibition of the Pin1-Smad3 interaction impacts TNBC cell migration/invasion, we treated cells with CYC065 or DMSO and assessed cell motility. CYC065 treatment significantly reduced the cells' ability to migrate and invade in all 3 TNBC cell lines (). Notably, Hs578T cells, which are the most migratory cell line in the study panel, were the most sensitive to CYC065 treatment. To investigate a mechanism behind the reduction of cell migration and invasion, we analyzed EMT-associated protein expression following CYC065 treatment and found a significant decrease in Snail, Slug, and β-catenin expression (). This was similar to the effect found after of Pin1 depletion ().
Figure 4. Inhibiting CDK-mediated Smad3 phosphorylation in TNBC cells reduces TNBC cell migration and invasion. Assays were performed 72 hours post-CYC065 treatment with (A) MDA-MB-231, (B) MDA-MB-436, and (C) Hs578T cells. Representative images were taken at 100X total magnification, and the relative number of migrated/invaded cells from at least 3 independent experiments is graphed. *p < 0.05, **p < 0.01, ***p < 0.001.
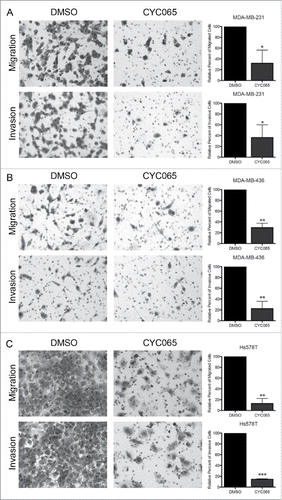
Figure 5. Inhibiting CDK-mediated Smad3 phosphorylation in TNBC cells hinders the EMT program. Expression of the EMT-associated proteins Snail, Slug, and β-catenin were quantified by immunoblot following CYC065 treatment with (A) MDA-MB-231, (B) MDA-MB-436, and (C) Hs578T cells. Snail and Slug expression was assessed 48 hours post-treatment and β-catenin was assessed after 72 hours. Representative immunoblots are shown with densitometry analysis of at least 3 independent experiments. *p < 0.05, **p < 0.01.
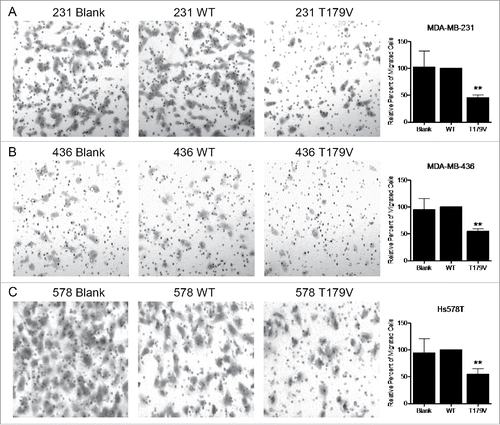
To further examine the impact of phosphorylation at the CDK2-target site T179, we virally transduced MDA-MB-231, MDA-MB-436, and Hs578T cells with an empty control vector, WT Smad3, or Smad3 phospho-mutant T179V. The T179V construct contains a single threonine to valine substitution at residue 179, thereby blocking the ability to be phosphorylated. Cells expressing WT Smad3 had similar motility as the control cells, however expression of the T179V construct significantly reduced the ability of TNBC cells to migrate through a transwell (). This is consistent with our previous finding that expression of the Smad3 5M construct, which has all 5 CDK2-target sites mutated, reduces TNBC cell migration.Citation18 This also suggests the importance of the T179 site specifically for the ability of cells to migrate.
Figure 6. Expression of a Smad3 mutant resistant to CDK-induced phosphorylation reduces TNBC cell migration. Migration assay performed with (A) MDA-MB-231, (B) MDA-MB-436, and (C) Hs578T cells that were transduced with lentivirus expressing an empty vector (Blank), wild type Smad3 (WT), or Smad3 with a T179V mutation (T179V). Representative images were taken at 100X total magnification, and the relative number of migrated cells from at least 3 independent experiments is graphed. **p < 0.01.
Pin1 negatively impacts Smad3 tumor-suppressive activity in TNBC cell lines
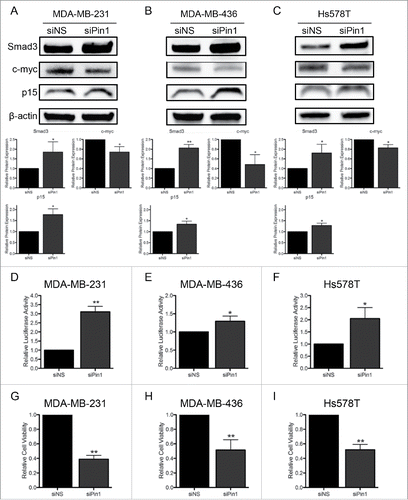
Next, we investigated the direct impact of the Pin1-Smad3 interaction on Smad3 function. The outcome of interaction with Pin1 varies by cell type, but often this interaction influences the stability of Pin1 targets.Citation23-25 Thus, we examined if interaction with Pin1 may target Smad3 for subsequent proteasomal degradation. KD of Pin1 with siPin1 in the 3 TNBC cell lines, MDA-MB-231, MDA-MB-436, and Hs578T, stabilized total Smad3 protein expression (, , ). As Smad3 functions as a transcription factor in the TGFβ pathway, we assessed transcription factor activity by luciferase assay. Cells were co-transfected with a construct containing a Smad3 responsive promoter upstream of the luciferase reporter (CAGA-luc) and a control construct containing the Renilla reporter. In accordance with the increase in total Smad3 levels, Pin1 KD promoted an increase in Smad3 transcription factor activity (, , ). Canonical tumor-suppressive Smad3 activity influences the cell cycle by promoting expression of the cdki p15 and inhibiting expression of c-myc.Citation27,28 Pin1 depletion contributed to a decrease in c-myc and an increase in p15 expression in all 3 cell lines (, , ). This correlated with a decrease in cell viability assessed by MTS assay (, , ). Taken together, the Pin1-Smad3 interaction is associated with pro-tumorigenic signaling in TNBC cells.
Figure 7. KD of Pin1 expression in TNBC cells stabilizes Smad3 protein expression and restores canonical Smad3 activity. Immunoblots of Smad3, c-myc, and p15 expression 96 hours post-Pin1 KD show an increase in Smad3 and p15 and a decrease in c-myc protein expression in (A) MDA-MB-231, (B) MDA-MB-436, and (C) Hs578T cells. Luciferase reporter assays of Smad3 activity 96 hours post-Pin1 KD were performed in (D) MDA-MB-231, (E) MDA-MB-436, and (F) Hs578T cells, and results of at least 3 independent experiments are graphed. A decrease in cell viability 96 hours post-Pin1 KD was quantified by MTS assay in (G) MDA-MB-231, (H) MDA-MB-436, and (I) Hs578T cells, and the results of at least 3 independent experiments are graphed. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Due to a current lack of targeted therapies, TNBC remains a highly aggressive breast cancer subtype associated with poor outcomes. In this study, we used 3 TNBC cell lines to explore a clinically applicable means of disrupting cancer cell migration/invasion and growth. It has previously been reported that Pin1, a peptidyl-prolyl cis/trans isomerase, recognizes and binds to the pT179 site in the linker region of Smad3.Citation20,22 We confirmed that Pin1 interacted with Smad3 in all the TNBC study cell lines. We found that inhibiting this interaction by depleting Pin1 greatly reduced TNBC cell migration and invasion, which corresponded to changes in EMT-associated proteins. Since CDK2 can phosphorylate the linker region of Smad3, including the T179 site, we treated cells with CYC065, a clinical stage CDK2/9 inhibitor. CYC065 treatment reduced Smad3 pT179 and inhibited the Pin1-Smad3 interaction. This corresponded to a decrease in cell migration/invasion following CYC065 treatment. Mutation of the pT179 site to inhibit phosphorylation by CDK2 also reduced cell migration. We found that depleting Pin1 expression restored Smad3 protein levels and canonical tumor-suppressive Smad3 activity. We conclude that CDK2-mediated phosphorylation of Smad3 promotes a Pin1-Smad3 interaction, which may lead to Smad3 proteasomal degradation and contributes to pro-tumorigenic and pro-metastatic Smad3/TGFβ pathway activity.
Pin1 is overexpressed in breast and other cancers, and this expression is linked to poor patient outcomes. In breast cancer, Pin1 expression is associated with increased tumor grade, cancer progression, and cancer aggressiveness.Citation23-25,29,30 Due to its clinical significance, numerous studies have sought to determine the mechanistic role of Pin1 in cancer. Pin1 expression has been linked with breast cancer stem cell expansion, potentially contributing to enhanced tumor growth and metastatic potential.Citation31,32 Depletion of Pin1 has also been found to reduce cell migration, invasion, and proliferation in breast and other cancer cell lines.Citation30,33-35 Matsuura et al. linked Pin1 to the Smad3/TGFβ pathway by demonstrating that Pin1 could interact with Smad3 and was associated with TGFβ-dependent prostate cancer cell migration and invasion.Citation20,22 Here, we provide evidence that Pin1 is necessary for the migration and invasion of TNBC cells. This aligns with previous studies demonstrating MDA-MD-231 cells with Pin1 KD had reduced cell migration in vitro and reduced mouse lung colonization following tail vein injection.Citation30
As a peptidyl-prolyl cis/trans isomerase, Pin1 recognizes and binds to phosphorylated serine/threonine-proline motifs of its targets. This includes the phosphorylated T179 site of Smad3.Citation20,22 T179 is located in the linker region of Smad3, and is a phosphorylation target of CDK2.Citation14,15 We found that inhibiting CDK2 with CYC065 decreased Smad3 pT179 and also reduced Pin1-Smad3 binding in the TNBC cell lines. CYC065 treatment also reduced TNBC cell migration and invasion, which aligns with previous studies implementing CDK2 or 4 inhibitory tool compounds to reduce Smad3 pT179 and TNBC migration and restore tumor-suppressive Smad3 activity.Citation16-18 We also found that mutation of the T179 site (Threonine to Valine mutation) reduced the motility of TNBC cells, underscoring the importance of CDK-mediated phosphorylation for TNBC cell migration. A recent study by Jo et al. also found that Smad3 pT179 was required for non-small lung cancer cell migration and EMT.Citation36 Since Pin1 is known to play a role in lung cancer carcinogenesis, future studies examining a potential Pin1-Smad3 interaction in lung cancer cells may be relevant.Citation34 Overall, our data show that the interaction of Pin1 and Smad3 is associated with TNBC cell migration/invasion: when the Pin1-Smad3 interaction was disrupted, either by depletion of Pin1 or inhibition of CDK2-mediated Smad3 phosphorylation, TNBC cell migration/invasion decreased.
In this current study, both CYC065 treatment and Pin1 depletion reduced expression of the transcription factors Snail and Slug, which are associated with TGFβ-induced EMT,Citation37,38 and β-catenin, a key player in the WNT pathway, which has a role in inducing EMT in breast cancer cells.Citation39 This suggests that CYC065 and Pin1 KD may impede the EMT process through a similar mechanism, potentially through blockade of the Pin1-Smad3 interaction, leading to a reduction in cell migration/invasion. Snail expression is downstream of the Smad3/TGFβ pathway, and the reduction in Snail could be due, in part, to the restoration of canonical tumor-suppressive Smad3 activity following CYC065 treatment or Pin1 KD.Citation40–42 The reduction of β-catenin aligns with a previous report showing that Pin1-deficient mice had decreased β-catenin expression. Additionally, Pin1 and β-catenin expression were correlated in human breast cancer tissues.Citation43 We found β-catenin expression was reduced at 48 hours post-CYC065 treatment, but the extent of reduction was greater at 72 hours. This may be secondary to Snail and Slug being upstream of the β-catenin/TCF-4 complex formation in the EMT signaling cascade. Down-regulation of Snail and Slug may produce a subsequent impairment of β-catenin stabilization and nuclearization.Citation44 It has also been shown that Snail can interact with β-catenin to promote Wnt signaling.Citation45 With a reduction in Snail expression following CYC065 treatment, Wnt signaling may decrease, leading to phosphorylation and degradation of β-catenin. Thus, a decrease in β-catenin expression may follow Snail reduction. Pin1 depletion resulted in an increase in Claudin, a tight junction protein associated with an epithelial cell phenotype. In breast cancer patients, low levels of claudin correlated with higher recurrence of disease.Citation46 These changes in EMT-associated protein expression suggest a mechanism for the decreased cell motility we observed following inhibition of the Pin1-Smad3 interaction.
The data in this report suggest that a pT179-dependent Pin1-Smad3 interaction plays a role in promoting the tumorigenic and metastatic action of non-canonical Smad3 signaling. We explored how Pin1 affected Smad3 activity and how this interaction may promote tumorigenesis. Interaction with Pin1 can influence function, localization and stability of the target protein.Citation24 Nakano et al. found overexpression of Pin1 induced a decrease in Smad3 protein levels, which could be rescued by treatment with a Pin1 inhibitor. Pin1 binding also enhanced the Smad3 interaction with the E3 ligase Smurf2 and ubiquitination of Smad3.Citation21 In accordance, we report that depletion of Pin1 restored Smad3 protein expression. We also found an increase in total Smad3 expression following CYC065 treatment, further supporting our hypothesis that inhibiting CDK2-mediated Smad3 phosphorylation prevents the inhibitory effects of the Pin1-Smad3 interaction. Pin1 depletion also restored canonical tumor-suppressive Smad3 activity. Under canonical tumor-suppressive TGFβ/Smad3 pathway signaling, Smad3 promotes the expression of tumor-suppressive genes, such as the cyclin dependent kinase inhibitor p15, and inhibits the expression of growth-promoting c-myc.Citation27,28 As such, we found that Smad3 transcriptional activity increased with Pin1 KD, which correlated with an increase in p15 expression and a decrease in c-myc expression. As a cdki, p15 controls cell growth by promoting cell cycle arrest.Citation28,47 In contrast, c-myc is a well-documented oncogene. Furthermore, an impact on CDK9 transcriptional action is a potential effect of CYC065 treatment. CDK9 phosphorylates and activates RNA polymerase II, and inhibition of CDK9 has also been shown to reduce transcription of oncogenes, including c-myc.Citation26,48 Farrell et al. analyzed breast cancer patient samples and found a co-overexpression of Pin1 and c-myc.Citation49 This pattern may be influenced by loss of tumor-suppressive Smad3 action. In accordance with these cell growth-related protein changes, we saw a decrease in cell viability following Pin1 depletion. In total, our results fit with prior findings identifying restoration of canonical Smad3 activity with inhibition of CDK-mediated phosphorylation.Citation16–18
The use of CDK inhibitors as a cancer therapy is an active area of study. CDKs are attractive targets because of their importance in cell cycle progression, which is a key process for the uncontrolled growth of cancer cells. However, many early CDK inhibitors were unsuccessful in clinical trials. This may have been due to lack of target specificity or absence of patient selection based on clinically relevant biomarkers.Citation11 With the recent development of CDK inhibitors with higher specificity, clinical successes have been identified. Accordingly, the FDA recently approved a CDK4/6 inhibitor, palbociclib, for ER+/HER- breast cancer patients. A phase II clinical study found that ER+ breast cancer patients had greater progression-free survival with combined palbociclib and letrozole therapy compared with letrozole alone.Citation50 Furthermore, recent work by Liu et al. identified a link between G1 cyclins/CDKs, Pin1, and the stability of stem cell factors. Depletion of cyclin D and E reduced stabilizing phosphorylation of Nanog, Oct4, and Sox2 and reduced their interaction with Pin1, suggesting that phosphorylation by cyclins/CDKs may promote Pin1 binding that protects proteins from proteasomal degradation. They also found that in TNBC cells, treatment with a CDK2i may target cancer ‘stem cell-like’ cells by reducing phosphorylation of and consequential protein stability of Sox2 and Oct4.Citation51 This suggests the need for additional studies examining the consequence of CDK2i treatment on tumor-initiating cell fate and the impact on tumor development and progression. Here, we examined the efficacy of a clinical CDK2/9 inhibitor, CYC065, in TNBC cells, as these cells can have cyclin E overexpression.Citation3-6 Moreover, our previous findings demonstrated a greater efficacy of CDK2i compared with CDK4i in TNBC xenograft tumor models.Citation3-6,18 CYC065 has also been implicated in causing cell death of HER2+ breast cancer cells with cyclin E-associated trastuzumab resistance.Citation8 Interestingly, Pin1 overexpression has also recently been linked to trastuzumab resistance, and inhibition of Pin1 may help sensitize resistant cells to treatment.Citation52 Further exploration of the relationship between Pin1, CDKs, and the Smad3/TGFβ pathway in HER2+ as well as triple negative disease may provide important insights into key mechanisms of treatment resistance for these aggressive breast cancer subtypes.
In this report, we outlined a molecular mechanism of CDK2 inhibition beyond canonical cell cycle regulation, which is crucial for the identification of potential biomarkers for CDK inhibitor therapy, including cyclin E and/or CDK2 overexpression, Smad3 linker region phosphorylation, and Pin1 overexpression. Future studies investigating Pin1 inhibitors alone and in combination with CDK2 inhibitors will also be pursued.Citation53 Our work adds to the mechanistic understanding of TGFβ-induced metastasis by providing greater insight into non-canonical CDK-mediated Smad3 signaling in TNBC, and enhances the experimental evidence supporting the use of CDK2 inhibitors for the treatment of TNBC.
Materials and methods
Cell lines, culture, and inhibitors
MDA-MB-231, MDA-MB-436, and Hs578T human TNBC cells, which are estrogen receptor (ER), progesterone receptor (PR), and Human Epidermal Growth Factor Receptor 2 (HER2) negative, were obtained from the American Type Culture Collection (ATCC). MDA-MB-231 and MDA-MB-436 cells were maintained in DMEM-F12 (Gibco, 11330057) and supplemented with non-essential amino acids, sodium pyruvate, antibiotics, glutamine, and 10% fetal bovine serum (FBS). Hs578T cells were maintained in DMEM (ATCC, 30–2002) supplemented with 0.01 mg/mL bovine insulin and 10% FBS. 293FT cells used for lentivirus creation were maintained in DMEM with high glucose (Gibco, 11965092) supplemented with non-essential amino acids, sodium pyruvate, antibiotics, glutamine, 10% FBS, and 500 µg/ml Geneticin (Gibco, A1113803). To pharmacologically inhibit CDK-mediated Smad3 phosphorylation, study cells were plated with 600nM CYC065 (Cyclacel). Control cells were treated with DMSO solvent alone.
RNA interference
MDA-MB-231, MDA-MB-436, and Hs578T study cells were seeded at 2.0 × 105 cells per well in a 6-well plate. After 24 hours, cells were transfected with Pin1 (siPin1) or non-specific control (siNS) small interfering RNA (ON-TARGETplus Smartpool siRNA; GE Dharmacon). Non-transfected cells were used as a control. Cells were incubated for 48 hours with the transfection mix containing 25 nM siRNA and 4 µL DharmaFECT Transfection Reagent (GE Dharmacon, T-2001–01) in serum free media (SFM). Transfected cells were collected and knock-down (KD) was confirmed by immunoblotting for Pin1 protein expression.
Lentiviral generation and transduction
To generate each of the viral mutants, 293FT cells were plated and co-transfected with 5 µg DNA constructs,Citation17 3 µg MDL-gag.pol, 1µg IVS-VSVG, and 1 µg RSV-rev (Shea Lab, University of Michigan, Ann Arbor, MI) using Lipofectamine 2000 (Invitrogen, 11668019). 293T cells were plated and allowed to grow to 90% confluence. The DNA constructs and the Lipofectamine 2000 were added to separate tubes containing OPTI-MEM (Gibco, 31985070) for 5 minutes. The contents of both tubes were mixed and incubated for 20 minutes at room temperature. One mL of the DNA/lipid complex was added to each plate and incubated at 37°C for 48 hours. The supernatant was collected, centrifuged for 5 minutes, and transferred to a new 50 mL tube. Ten mL of 5X PEG-it solution (System Biosciences, LV810A-1) was added, and the mix was incubated at 4°C for 24 hours. The tube was then centrifuged and the viral pellet re-suspended in PBS, aliquoted, and stored at −80°C. To transduce the study cells, 2.0 × 105 cells per well were plated in 6-well plates and allowed to grow overnight. Cells were incubated at 37°C with 10 µL of viral solution for 24 hours. Successfully transduced cells were selected with puromycin and maintained in the same fashion as the parental cell lines with the addition of puromycin (2 µg/ml for 231 cells, 5 µg/ml for 436 cells, and 1 µg/ml for 578 cells) to the media. Expression of the constructs was confirmed by immunoblotting for total Smad3 expression.
Immunoblotting and antibodies
Following 48-hour treatment with DMSO/CYC065 or Pin1 KD (96 hours post-siPin1/siNS transfection), study cells were lysed with M-PER reagent (Thermo Scientific, 78503) containing protease and phosphatase inhibitors (CST, 5872). Protein was quantified with Bradford reagent (Bio-Rad, 5000006). Twenty µg of lysate was mixed with 2X Laemmli sample buffer (Bio-Rad 161–0737) containing 5% 2-Mercaptoethanol (Bio-Rad, 161–0710), loaded into precast 4–15% polyacrylamide gels (Bio-Rad, 4561084), and electrophoresed. Protein was transferred to nitrocellulose membranes, blocked in BLOK™ casein (G Biosciences, 786–196) or tris-buffered saline with 0.1% Tween-20 (TBST-T) containing 5% bovine serum albumin (BSA) for one hour, and incubated with primary antibody in casein or BSA overnight at 4°C. After washing, the membrane was incubated with diluted secondary antibody for one hour, exposed to ECL reagent (Bio-Rad, 1705060) for 5 minutes, and visualized with a ChemiDoc MP Bio-Rad imager. For re-blotting, membranes were stripped of antibody with stripping buffer (Thermo Scientific, 21059) and washed before re-probing. Image analysis was performed with Bio-Rad Image Lab software.
The following antibodies were used for western blotting: rabbit monoclonal to β-Actin (CST, 4970), rabbit monoclonal to β-catenin (CST, 8480), rabbit monoclonal to Claudin (CST, 13255), rabbit polyclonal p15/INK4B (Abcam, ab53034), rabbit polyclonal to Pin1 (CST, 3722), rabbit monoclonal c-myc (Abcam, ab32072), rabbit monoclonal to Slug (CST, 9585), rabbit polyclonal Smad3 (Abcam, ab28379), rabbit monoclonal Snail (CST, 3879). The primary rabbit p-Smad3-T179 antibody was a generous gift from Dr. Fang Liu (Rutgers University, New Jersey). Additional antibodies used for coIPs: mouse monoclonal to Pin1 (R&D Systems, MAB2294), normal mouse IgG (Santa Cruz, sc-2025), and rabbit polyclonal Smad3 (Invitrogen, 511500).
Cell migration and invasion assays
MDA-MB-231, MDA-MB-436, and Hs578T cells were: transfected with siPin1 or siNS; treated with CYC065 or DMSO; or transduced with B, WT, or T179V lentiviral constructs as described above. Cells were serum starved for 24 hours. Matrigel-coated or uncoated transwells with highly porous 8 μm polyethylene terephthalate membranes (Corning, 354483, 35309) were placed in the wells of a 24-well plate for invasion or migration assays, respectively. Wells also contained complete media with 10% FBS as a chemoattractant or SFM as a negative control. In each transwell, 50,000 serum-starved cells were plated in SFM in triplicate and allowed to invade and migrate. The inserts were removed and fixed with 0.5% crystal violet. Images were captured at 100X magnification and the number of invaded/migrated cells was counted using ImageJ software.
Immunoprecipitation assays
Study cells were treated with DMSO control or CYC065 for 48–72 hours, lysed, and protein was collected for analysis using a co-immunoprecipitation kit (Pierce, 26149) following the manufacturer's directions. Briefly, 1–2 mg of protein lysate was pre-cleared with control resin for 1 hour at 4°C, and incubated with Pin1 antibody-immobilized resin overnight at 4°C. The beads were washed and the bound protein eluted. Elute was analyzed by immunoblotting for total Smad3 and Pin1.
Luciferase assays
Smad3 reporter plasmid (CAGA) and control plasmid (Renilla) were co-transfected into study cells 48 hours post-Pin1 KD. To control for background luminescence, a portion of cells were not transfected. Smad3 activity was measured with the Dual Luciferase Reporter Assay System (Promega, E1910). Briefly, cells were lysed with passive lysis buffer 48 hours post-transfection and lysate was added to a 96-well plate in triplicate. Luminescence was read with a BioTek Synergy 2 plate reader, which injected luciferase and renilla substrates and measured output, respectively. Fluc signal was normalized to Rluc signal.
MTS assays
Study cells were plated in a 96-well plate in triplicate 48 hours post-Pin1 KD. They were allowed to grow for 48 hours (96 hours post-KD). CellTiter 96 AQ One Solution Reagent (Promega, G3580) reagent was added to each well and absorbance was measured at 490nm with a Biotek ELx800 plate reader.
Statistics
Data are presented as mean ± standard deviation. In general, data were normalized to the control. To determine significant differences between treatments/conditions, paired 2-tailed t tests were performed. p-values less than 0.05 were considered significant. In all cases, at least 3 experimental replicates were performed.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. David Blake and Dr. Daniella Zheleva (Cyclacel Ltd.) for generously providing CYC065, and for helpful discussions regarding the dosage requirements for CYC065.
Funding
The authors acknowledge financial support from the Society of Surgical Oncology - Clinical Investigator Award (to J.S. Jeruss) and NIH R01GM097220 (to J.S. Jeruss), A Sister's Hope Foundation, and the Saslow Family (J.S.Jeruss). Alexandra Thomas is supported by NIH 5F31CA206259–02.
References
- Guarneri V, Broglio K, Kau SW, Cristofanilli M, Buzdar AU, Valero V, Buchholz T, Meric F, Middleton L, Hortobagyi GN, et al. Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors. J Clin Oncol 2006; 24:1037-44; PMID:16505422; https://doi.org/10.1200/JCO.2005.02.6914
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010; 363:1938-48; PMID:21067385; https://doi.org/10.1056/NEJMra1001389
- Nielsen NH, Arnerlov C, Emdin SO, Landberg G. Cyclin E overexpression, a negative prognostic factor in breast cancer with strong correlation to oestrogen receptor status. Br J Cancer 1996; 74:874-80; PMID:8826852; https://doi.org/10.1038/bjc.1996.451
- Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, Bedrosian I, Knickerbocker C, Toyofuku W, Lowe M, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med 2002; 347:1566-75; PMID:12432043; https://doi.org/10.1056/NEJMoa021153
- Fadare O, Tavassoli FA. Clinical and pathologic aspects of basal-like breast cancers. Nat Clin Practice Oncol 2008; 5:149-59; PMID:18212769; https://doi.org/10.1038/ncponc1038
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature 2000; 406:747-52; PMID:10963602; https://doi.org/10.1038/35021093
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001; 411:1017-21; PMID:11429595; https://doi.org/10.1038/35082500
- Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, Chandarlapaty S, Serra V, Prat A, Ibrahim YH, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci U S A 2011; 108:3761-6; PMID:21321214; https://doi.org/10.1073/pnas.1014835108
- Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, Bazarov AV, Smyth JW, Davis SE, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med 2012; 209:679-96; PMID:22430491; https://doi.org/10.1084/jem.20111512
- Migliaccio I, Di Leo A, Malorni L. Cyclin-dependent kinase 4/6 inhibitors in breast cancer therapy. Curr Opin Oncol 2014; 26:568-75; PMID:25188473; https://doi.org/10.1097/CCO.0000000000000129
- Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discovery 2015; 14:130-46; PMID:25633797; https://doi.org/10.1038/nrd4504
- Aleem E, Berthet C, Kaldis P. Cdk2 as a master of S phase entry: fact or fake? Cell Cycle 2004; 3:35-7; PMID:14657662; https://doi.org/10.4161/cc.3.1.632
- Berthet C, Klarmann KD, Hilton MB, Suh HC, Keller JR, Kiyokawa H, Kaldis P. Combined loss of Cdk2 and Cdk4 results in embryonic lethality and Rb hypophosphorylation. Dev Cell 2006; 10:563-73; PMID:16678773; https://doi.org/10.1016/j.devcel.2006.03.004
- Sekimoto G, Matsuzaki K, Yoshida K, Mori S, Murata M, Seki T, Matsui H, Fujisawa J, Okazaki K. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res 2007; 67:5090-6; PMID:17545585; https://doi.org/10.1158/0008-5472.CAN-06-4629
- Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004; 430:226-31; PMID:15241418; https://doi.org/10.1038/nature02650
- Cooley A, Zelivianski S, Jeruss JS. Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines. Cell Cycle 2010; 9:4900-7; PMID:21150326; https://doi.org/10.4161/cc.9.24.14158
- Zelivianski S, Cooley A, Kall R, Jeruss JS. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol Cancer Res 2010; 8:1375-87; PMID:20736297; https://doi.org/10.1158/1541-7786.MCR-09-0537
- Tarasewicz E, Rivas L, Hamdan R, Dokic D, Parimi V, Bernabe BP, Thomas A, Shea LD, Jeruss JS. Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells. Cell Cycle 2014; 13:3191-201; PMID:25485498; https://doi.org/10.4161/15384101.2014.950126
- Gao S, Alarcon C, Sapkota G, Rahman S, Chen PY, Goerner N, Macias MJ, Erdjument-Bromage H, Tempst P, Massague J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol Cell 2009; 36:457-68; PMID:19917253; https://doi.org/10.1016/j.molcel.2009.09.043
- Aragon E, Goerner N, Zaromytidou AI, Xi Q, Escobedo A, Massague J, Macias MJ. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev 2011; 25:1275-88; PMID:21685363; https://doi.org/10.1101/gad.2060811
- Nakano A, Koinuma D, Miyazawa K, Uchida T, Saitoh M, Kawabata M, Hanai J, Akiyama H, Abe M, Miyazono K, et al. Pin1 down-regulates transforming growth factor-beta (TGF-beta) signaling by inducing degradation of Smad proteins. J Biol Chem 2009; 284:6109-15; PMID:19122240; https://doi.org/10.1074/jbc.M804659200
- Matsuura I, Chiang KN, Lai CY, He D, Wang G, Ramkumar R, Uchida T, Ryo A, Lu K, Liu F. Pin1 promotes transforming growth factor-beta-induced migration and invasion. J Biol Chem 2010; 285:1754-64; PMID:19920136; https://doi.org/10.1074/jbc.M109.063826
- Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J 2001; 20:3459-72; PMID:11432833; https://doi.org/10.1093/emboj/20.13.3459
- Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 2007; 8:904-16; PMID:17878917; https://doi.org/10.1038/nrm2261
- Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol 2004; 164:1727-37; PMID:15111319; https://doi.org/10.1016/S0002-9440(10)63731-5
- Blake DG, MacKay C, Frame S, Zheleva D. CYC065, a novel CDK2/9 inhibitor: Molecular basis for clinical development in basal-like triple-negative breast cancer. Cancer Res 2016; 76(4 Suppl):Abstract nr P5-03-10; https://doi.org/10.1158/1538-7445.SABCS15-P5-03-10
- Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 2002; 110:19-32; PMID:12150994; https://doi.org/10.1016/S0092-8674(02)00801-2
- Feng XH, Liang YY, Liang M, Zhai W, Lin X. Direct interaction of c-Myc with Smad2 and Smad3 to inhibit TGF-beta-mediated induction of the CDK inhibitor p15(Ink4B). Mol Cell 2002; 9:133-43; PMID:11804592; https://doi.org/10.1016/S1097-2765(01)00430-0
- Rustighi A, Zannini A, Campaner E, Ciani Y, Piazza S, Del Sal G. PIN1 in breast development and cancer: a clinical perspective. Cell Death Differ 2017; 24:200-11; PMID:27834957; https://doi.org/10.1038/cdd.2016.122
- Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A, et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011; 20:79-91; PMID:21741598; https://doi.org/10.1016/j.ccr.2011.06.004
- Rustighi A, Zannini A, Tiberi L, Sommaggio R, Piazza S, Sorrentino G, Nuzzo S, Tuscano A, Eterno V, Benvenuti F, et al. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol Med 2014; 6:99-119; PMID:24357640; https://doi.org/10.1002/emmm.201302909
- Luo ML, Gong C, Chen CH, Lee DY, Hu H, Huang P, Yao Y, Guo W, Reinhardt F, Wulf G, et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer Res 2014; 74:3603-16; PMID:24786790; https://doi.org/10.1158/0008-5472.CAN-13-2785
- Ryo A, Uemura H, Ishiguro H, Saitoh T, Yamaguchi A, Perrem K, Kubota Y, Lu KP, Aoki I. Stable suppression of tumorigenicity by Pin1-targeted RNA interference in prostate cancer. Clin Cancer Res 2005; 11:7523-31; PMID:16243827; https://doi.org/10.1158/1078-0432.CCR-05-0457
- Tan X, Zhou F, Wan J, Hang J, Chen Z, Li B, Zhang C, Shao K, Jiang P, Shi S, et al. Pin1 expression contributes to lung cancer: Prognosis and carcinogenesis. Cancer Biol Therapy 2010; 9:111-9; https://doi.org/10.4161/cbt.9.2.10341
- Atabay KD, Yildiz MT, Avsar T, Karabay A, Kilic T. Knockdown of Pin1 leads to reduced angiogenic potential and tumorigenicity in glioblastoma cells. Oncol Letters 2015; 10:2385-9; PMID:26622856
- Jo E, Park SJ, Choi YS, Jeon WK, Kim BC. Kaempferol Suppresses Transforming Growth Factor-beta1-Induced Epithelial-to-Mesenchymal Transition and Migration of A549 Lung Cancer Cells by Inhibiting Akt1-Mediated Phosphorylation of Smad3 at Threonine-179. Neoplasia 2015; 17:525-37; PMID:26297431; https://doi.org/10.1016/j.neo.2015.06.004
- Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS One 2011; 6:e26514; PMID:22028892; https://doi.org/10.1371/journal.pone.0026514
- Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res 2009; 19:156-72; PMID:19153598; https://doi.org/10.1038/cr.2009.5
- Wang Y, Zhou BP. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chinese J Cancer 2011; 30:603-11; PMID:21880181; https://doi.org/10.5732/cjc.011.10226
- Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem 2003; 278:21113-23; PMID:12665527; https://doi.org/10.1074/jbc.M211304200
- Dzwonek J, Preobrazhenska O, Cazzola S, Conidi A, Schellens A, van Dinther M, Stubbs A, Klippel A, Huylebroeck D, ten Dijke P, et al. Smad3 is a key nonredundant mediator of transforming growth factor beta signaling in Nme mouse mammary epithelial cells. Mol Cancer Res 2009; 7:1342-53; PMID:19671686; https://doi.org/10.1158/1541-7786.MCR-08-0558
- Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol 2009; 11:943-50; PMID:19597490; https://doi.org/10.1038/ncb1905
- Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol 2001; 3:793-801; PMID:11533658; https://doi.org/10.1038/ncb0901-793
- Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell 2008; 19:4875-87; PMID:18799618; https://doi.org/10.1091/mbc.E08-05-0506
- Stemmer V, de Craene B, Berx G, Behrens J. Snail promotes Wnt target gene expression and interacts with beta-catenin. Oncogene 2008; 27:5075-80; PMID:18469861; https://doi.org/10.1038/onc.2008.140
- Morohashi S, Kusumi T, Sato F, Odagiri H, Chiba H, Yoshihara S, Hakamada K, Sasaki M, Kijima H. Decreased expression of claudin-1 correlates with recurrence status in breast cancer. Int J Mol Med 2007; 20:139-43; PMID:17611630
- Roussel MF. The INK4 family of cell cycle inhibitors in cancer. Oncogene 1999; 18:5311-7; PMID:10498883; https://doi.org/10.1038/sj.onc.1202998
- Sengupta S, Biarnes MC, Jordan VC. Cyclin dependent kinase-9 mediated transcriptional de-regulation of cMYC as a critical determinant of endocrine-therapy resistance in breast cancers. Breast Cancer Res Treatment 2014; 143:113-24; PMID:24309997; https://doi.org/10.1007/s10549-013-2789-2
- Farrell AS, Pelz C, Wang X, Daniel CJ, Wang Z, Su Y, Janghorban M, Zhang X, Morgan C, Impey S, et al. Pin1 regulates the dynamics of c-Myc DNA binding to facilitate target gene regulation and oncogenesis. Mol Cell Biol 2013; 33:2930-49; PMID:23716601; https://doi.org/10.1128/MCB.01455-12
- Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, Zirkelbach JF, Yu J, Liu Q, Zhao L, et al. FDA Approval: Palbociclib for the treatment of Postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res 2015; 21:4760-6; PMID:26324739; https://doi.org/10.1158/1078-0432.CCR-15-1185
- Liu L, Michowski W, Inuzuka H, Shimizu K, Nihira NT, Chick JM, Li N, Geng Y, Meng AY, Ordureau A, et al. G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nat Cell Biol 2017; 19:177-88; PMID:28192421; https://doi.org/10.1038/ncb3474
- Sajadimajd S, Yazdanparast R. Sensitizing effect of juglone is mediated by down regulation of Notch1 signaling pathway in trastuzumab-resistant SKBR3 cells. Apoptosis 2017; 22:135-44; PMID:27770268; https://doi.org/10.1007/s10495-016-1291-9
- Min SH, Zhou XZ, Lu KP. The role of Pin1 in the development and treatment of cancer. Arch Pharmacal Res 2016; 39:1609-20; PMID:27572155; https://doi.org/10.1007/s12272-016-0821-x