
ABSTRACT
Estrogen mediates various cellular processes including cell proliferation, differentiation, growth and mammary gland function. Estrogen Receptors (ERs) are expressed in 70% of breast cancers. Consequently, estrogen mediated ER signaling plays a critical role in breast cancer diagnosis, prognosis, and treatment. ERs are ligand-triggered transcription factors. However, in the absence of a cognate estrogenic ligand, ERs can be activated by a variety of other extracellular signals. Tamoxifen, an anti-estrogen that selectively targets ER, induces substantial regression of breast tumors and an increase in disease-free survival. Tamoxifen mimics estrogen effects in other tissues thereby providing some beneficial effects including reduced risk of osteoporosis. However, breast cancers that initially respond well to tamoxifen tend to develop resistance and resume growth despite the continued presence of the antagonist.
Library of compounds with substituted morpholinoaniline scaffold, a set of structurally divergent potential ER antagonists that fit the tamoxifen pharmacophore, were designed to target ER Ligand Binding Domain (LBD) and to recruit co-regulator proteins including BRCA1 over a range of conformational changes. Two of the lead compounds in the library, BR46 and BR47, were found to inhibit estrogen induced cell proliferation and cell viability. Discovery of novel lead molecules targeting ligand binding pockets of hER has provided structural clues toward the development of new breed of small molecule therapeutics for tamoxifen-resistant breast cancers and would complement already existent anti-estrogen therapy.
Introduction
Breast cancer is the second leading cause of deaths due to cancer. The American Cancer Society predicts that there will be 252,710 new cases of invasive breast cancer diagnosed and approximately 40,610 women will die from breast cancer in 2017 (Americal Chemical Society Report, 2017). The female hormone estrogen (17-β-estradiol) has been found to promote the advancement of breast cancer. Estrogen enters the cell through passive diffusion and elicits both genomic and nongenomic cell signaling by binding to a host of protein targets. Estrogen Receptor (ER), the primary target for estrogen, is a nuclear hormone receptor that is activated upon binding its endogenous hormone estrogen. Intracellular ERs, ER α (encoded in chromosome 6) and ER β (encoded in chromosome 14) are made up of 3 major domains termed the activation function one (AF-1), DNA binding domain (DBD) and the activation function 2/ligand binding domain (AF-2/LBD), which are linked through less organized hinge regions.Citation1,2 Estrogen receptor α and β share 58% and 95% sequence similarity in the LBD and DBD respectively.Citation3-5 The LBD is composed of 12 helices and 2 β sheets connected through loops.Citation6,23 The AF2 domain is widely reported to be important for the ER/coactivator interactions,Citation7-10 the AF1 domain in facilitating the ER/transcription factor interactionCitation11 and the hinge region for receptor dimerization.Citation12 The classical ER model of transactivation involves a cascade of conformational changes post estrogen binding which allows ER dimerization, which then translocates to the nucleus, binds to estrogen response elements (ERE), recruits coregulator proteins/nuclear transcriptional machinery and ultimately inducing transcription of various target genes, such as ps2 and progesterone receptor.Citation13 In addition, ER can mediate transcription at other response elements through protein-protein interaction, as seen with the activator protein 1 transcription factor complex (AP-1) composed of Fos and Jun proteins. In fact, one third of the genes regulated by ER do not contain typical ERE.Citation14 The full length ER is present in more than 70% of breast cancers and overexpressed in 50% of breast cancers and deleterious estrogen signaling can lead to breast cancer progression.Citation15 Therefore ER is one of the primary chemotherapeutic targets, and anti-estrogens such as tamoxifen have been on the market as anti-cancer agents for many years. However 30% of ER positive tumors are initially resistant and others gain acquired resistance over time.Citation16
Estrogen receptor modulators
In addition to endogenous estrogens, other small molecules have been shown to bind and regulate ERs. Ever since Lerner's group described the anti-estrogenic activity of ethamoxytriphetol,Citation17 there is a consistent interest in the discovery of selective and high affinity small molecular ligands targeting estrogen receptor. This resulted in 2 primary classes of anti-estrogens (i) selective estrogen receptor modulators (SERMs) and (ii) selective estrogen receptor downregulators (SERDs).Citation18 SERMs are classified by the divergent activity they can induce depending on the tissue context, and can be subdivided into 2 categories which both describe non-steroidal compounds, “tamoxifen-like” (triphenylethylenes) and “fixed ring” compounds. SERDs vary from SERMs in that they elicit purely antagonist activity despite the tissue type and are called pure antagonist accordingly.
Tamoxifen thus far has been the most successful SERM as an adjuvant breast cancer therapy and chemopreventive agent. It is responsible for saving approximately 500,000 women's lives.Citation19 Tamoxifen acts as an antagonist in the breast, but an agonist in the uterus. The differential biological activity of ER targeting small molecules is thought to be dependent on coregulator expression levels, ER posttranslational modifications and ER subtype expression levels.Citation20 Prolonged treatment with tamoxifen could lead to increased risk of endometrium cancer.Citation21 In addition to tamoxifen, the triphenylethylene SERM class of compounds includes: nitromifene, clomiphene, toremifene, droloxifene, TAT-59 and idoxifene, which vary from tamoxifen by a change in side chain.Citation22-26 The “fixed ring” subclass includes: raloxifene; a benzothiophene derivative, arzoxifene ERA-923, nafoxidine, trioxifene, a benzopyran derivative, and EM-800; a benzopyran derivative.Citation27-30 The SERD class of compounds includes: fulvestrant (ICI182780), SR 16234 and ZK 191703. Fulvestrant, the first SERD to enter clinical trials,Citation18 was found to inhibit tamoxifen resistant breast cancers in vitro and in vivo, lacking cross resistance with tamoxifen.Citation31-34 The broad pharmacological profile of fulvestrant is distinct from tamoxifen, in that fulvestrant is a more potent antagonist in vitro and in vivo, delays the onset of tumor growth for twice as long as tamoxifen, and does not have uterine-stimulating activity.Citation35,36
SERM-ER complexes still dissociate from heat shock proteins in the cytosol, dimerize, translocate to the nucleus, bind DNA and recruit coregulator proteins. While SERD-ER complexes dissociate from heat shock proteins, they are unable to form dimers and translocate to the nucleus.Citation37 Consequently, the ER accumulation in the cytosol results in an increase in ER degradation. Moreover, tamoxifen inhibits ER by binding to the AF2 domain, while fulvestrant antagonizes ER by binding to both the AF1 and AF2 domain.Citation38-41 SERDs downregulates ERα protein levels through hyperubiquitination and the proteasomal degradation pathway.Citation42 Many SERDs are steroidal analogs of estrogen and have bulky side chains at the 7α and 11β position such as ZD 164384 and RU 58668 respectively.Citation43 However, the limited oral bioavailability remains a problem as seen in fulvestrant. More recently several SERD compounds have been developed that show therapeutic promise based on the cyclofenil and bicyclononane scaffolds.Citation44
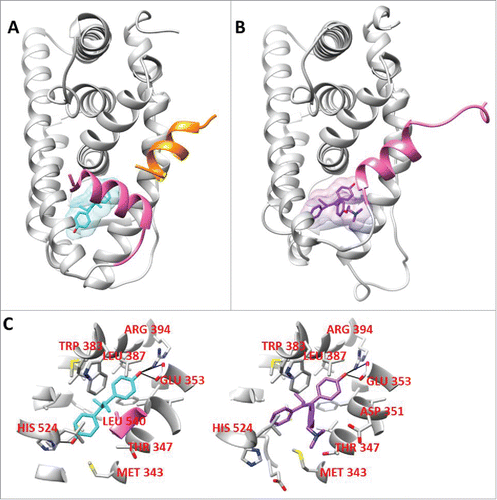
The agonist conformation of ER is highlighted by the positioning of helix 12 over a hydrophobic pocket against helices 3 and 11, thereby allowing coactivator recruitment.Citation6 The antagonist conformation of ER is highlighted by the distinct orientation helix 12 is induced by an antagonist ligand overlapping the co-activator binding pocket thereby blocking its recruitment (). Synthetic estrogen diethyl stilbesterol (DES; agonist) and 4-hydroxy tamoxifen (4OHT; antagonist) binds to the estrogen receptor through a high affinity interaction driven by 3 hydrogen bond interactions with glutamic acid 353, arginine 394, and histidine 524 of ERα (). These changes in helix 12 lead to a change in the shape of the coregulator binding surface of ER.Citation45-50 Interestingly, ER D351Y is a naturally occurring mutant isolated from a MCF7 cell line that was continually passagedCitation51,52 was found to enhance the estrogenic activity of tamoxifen.Citation53 Moreover, another challenge of hormone therapy is initial or acquired resistance. The exact causes of this resistance are unknown, however, a variety of theories exists including acquired ER mutations, posttranslational modifications or differential coregulator expression.Citation54
Figure 1. A. An agonist conformation of ER (PDB ID: 3ERD) co-crystallized with diethylstilbestrol; B. An antagonist conformation of ER (3ERT) co-crystallized with 4-hydroxytamoxifen; C. The ER residues that make up the 5 Å zone around the prospective compounds co-crystallized with the LBD of ER, their hydrogen bonding interactions are highlighted in black.
There is a significant need to identify new anti-estrogens that could potentially exploit increased interactions with known ER mutations, post-translational modifications and enhanced co-repressor recruitment. Here we describe, the design and discovery of novel anti-estrogens with morpholinoaniline scaffold and evaluate their role as potential anti-proliferative agents in human breast cancer cells in vitro.
Materials and methods
Chemicals All solvents and compounds purchased were analytical reagent grade. 2,5-diethoxy-4-morpholinoaniline, 4-morpholinoaniline, 2-methoxy-4-morpholinoaniline, diisopropylethylamine, potassium carbonate, sodium ascorbate, copper (II) sulfate, bromoacetic acid, propargyl bromide, azidopentanoic acid, 4-azidophenacyl bromide, 4-azidoaniline hydrochloride, sodium azide, 2-bromoethanol, 3-bromopropanol, 4-bromobutanol, 5-bromopentanol, epibromohydrin, acetone, chloroform, methanol, acetonitrile, ethyl acetate, deuterium oxide, and deuterated chloroform were purchased from Sigma-Aldrich.
Small Molecule Structure-property Analysis and Molecular Docking Each compound's 2 and 3 dimensional coordinates were generated using Symx draw and Marvin sketch respectively. ChemMine Tools was used to compare structure-physicochemical property of the compound library and with estrogenic reference ligands.Citation55 This includes comprehensive structure based clustering using atom pair similarity. Atom pair structural descriptors were used to determine the similarity, and then used in generating a similarity coefficient between 2 compounds by enumerating the common and unique structural and chemical properties.Citation55 The compounds were then subjected to in silico docking against 4 conformationally distinct X-ray crystal structures (Protein data bank (PDB)Citation56 ID: 1ERR, 3ERD, 1QKM, and 1U3S) of the ligand binding domain of human estrogen receptor (UniProt ID: P03372).Citation57 Autodock (The Scripps Research Institute) and SYBYL X (Tripos, St. Louis, MO) were used to dock the library. The ligands were docked in reference to the crystal structure bound ligand as a reference and rank ordered based on their docking scores. Briefly, potential ligand binding sites (ProtoMol) were defined for each protein-ligand complex based on the reference ligand bound in the original crystal structure. The protomol_bloat was set to zero and the protomol_threshold was set to 0.50 Å, and all other parameters were set to default values. Ligands were rendered flexible in surflex docking routine, multiple conformations (docked poses) were generated and scored using surflex score (CScore) which includes a combination of Dock-score, Gold-score, PMF-score and CHEM-score. Twenty conformations for each protein-ligand docking experiement were retrieved and top docked poses were selected based on the orientation of the test ligand with respect to the reference ligand and its CScore value.
Instrumentation Mass spectra data were collected with an Agilent Liquid Chomatograph (1200 series) Mass Spectrometer quadrupole (model G6130A) equipped with a ZORBAX Eclipse XDB-C18 column (Agilent) was used to confirm the synthesized compounds by using the following method: fragmentor voltage of 100 V, method spray chamber was set to API-ES, drying gas flow of 9 Ls/minute, nebulizer pressure of 40 psig, drying gas temperature of 300°C, capillary voltage of 3000 V, mobile phase was 65% methanol, 34.8% H2O and 0.2% trifluoroacetic acid, run time of 15–20 minutes, positive polarity and scan mode. Proton NMR spectra were collected with a 400 MHz Varian 400-MR nuclear magnetic resonance spectrometer and analyzed with the vnmrj software.
Chemical Synthesis The lead compounds BR 46 and BR47 were synthesized by adding 2,5-diethoxy-4-morpholinoaniline (1mmol, 340mg) to a stirring mixture of diisopropylethylamine (0.43mmol, 10ml), and 4-bromobutanol (10mmol, 900 µl) or 5-bromopentanol (10mmol, 1 ml) in 25ml of chloroform. The reaction was performed overnight under reflux and the reaction progress was monitored with thin layer chromatography (TLC; chloroform/methanol 95%/5%). The final product was purified by column chromatography (silica gel, chloroform/methanol 9/1). The identity and purity of the final products were confirmed with mass spectroscopy and Proton NMR (in deuterated chloroform).
Cell Culture MCF-7 and MDA-MB-231 breast cancer cells were originally obtained from the American Type Culture Collection (ATCC) and were cultured according to the suggested ATCC protocol as follows (https://www.atcc.org/∼/ps/HTB-22.ashx): 4 × 104 cells were seeded in a T-75 vented flask and grown to about 70% confluency in Minimum Essential Medium (MEM, https://www.thermofisher.com/us/en/home/life-science/cell-culture/mammalian-cell-culture/classical-media/mem.html) supplemented with 10% fetal bovine serum (http://cellgro.com/products/serum/animal-sera/fetal-bovine-serum-regular.html or http://cellgro.com/charcoal-stripped-fbs-premium-quality.html before treatments), 0.01 mg/ml of human recombinant insulin (https://www.thermofisher.com/us/en/home/life-science/bioproduction/bp-misc/recombinant-human-insulin-reagent.html) and 5% penicillin streptomycin (http://cellgro.com/products/microbiology-and-molecular-biology/antibiotics-antimycotics/penicillin-streptomycin-solution-100x.html). Cells were housed at 37°C with 5% CO2 and 10% humidity. Two days before the treatments cells were changed to phenol-free MEM media (https://www.thermofisher.com/order/catalog/product/51200038).
Cell viability A seed density of 80 × 104 cells was added to each well of a clear 96 well plate (Corning) in in phenol-free MEM supplemented media, as described previously.Citation58 Briefly, the cells were incubated overnight at 37°C with 5% CO2 and 10% humidity. The cells were treated with 0.05 to 100 uM of test compounds alone or in the presence of 1nM E2. Control wells were treated with 0.5% DMSO and 4OHT was used as vehicle and control respectively. Each experimental condition was done in triplicates and at least 2 independent experiments. The cells were treated for 12, 24, 48 hours at 37°C with 5% CO2 and 10% humidity. Thiazolyl bromide (1 mM) was added to each well, excluding negative control wells, to which phenol-free MEM supplemented media was added. The cells were incubated with thazolyl bromide for 4 hours and the absorbance at 570 nm and 650 nm was monitored using the Bio-Tek Synergy microplate reader.
Cell proliferation was assessed using a fluorescent ethynyldeoxyuridine (EdU) incorporation or Click-iT assay (https://www.thermofisher.com/order/catalog/product/C10337). All reagents and assay plates provided by the manufacturer were used as described previously.Citation58 Briefly, approximately 80 × 102 cells in phenol-free MEM supplemented media was added to each well of a 96 well black side, clear bottom plate. The cells were incubated overnight at 37°C with 5% CO2 and 10% humidity. The cells were treated with 5 uM of test compounds and/or 1 nM E2. Control wells were treated with 0.1% DMSO. Each experimental condition was done in triplicates and at least 3 independent experiments. The cells were treated for 8 hours at 37°C with 5% CO2 and 10% humidity. 10uM EdU (5-ethynyl-2′-deoxyuridine) was added to each well, except for negative control wells in which media alone was added instead. The cells were incubated with EdU for 1 hour at 37°C with 5% CO2 and 10% humidity. The medium was then removed, and the cells were fixed with 3.7% formaldehyde in PBS for 15 minutes at room temperature and washed with PBS twice. Next the cells were permeabilized with 0.1% Triton-X 100 in PBS for 15 minutes at room temperature and washed twice with PBS. EdU incorporation was detected using a Click-iT reaction. The Click-iT reaction mixture was prepared according to the protocol suggested by Life Technologies and the cocktail contained CuSO4 and Alexa Fluor 488 Azide (5-Carboxamido-(6-Azidohexanyl), Bis(Triethylammonium Salt)), 5-siomer). The Click-iT cocktail was incubated with cells for 30 mins at room temperature in the dark. After the cocktail was removed the cells were washed with Click-iT reaction rinse buffer. HCS NuclearMask Blue stain (diluted 1:2000 in PBS) was then incubated with the cells for 30 minutes at room temperature in the dark. The cells were washed twice with PBS. The fluorescence of each well was then quantified with the Bio-Tek Synergy microplate reader using the 495 nm excitation and 519 nm emission filters and representative images of each well were captured with the Zeiss Axio Observer Inverted Microscope. Images were analyzed using Image J software.
Flow Cytometry A seed density of 5 × 105 was added to T-25 vented flask in MEM supplemented medium and allowed to grow for 70–80% confluency. Before the cells were treated the growth medium was removed and fresh supplemented DMEM, as described previously,Citation58 was added to each flask. The cells were then treated with 5 uM test compounds and/or 1 nM E2 or 1 uM 4OHT for 16 hours or 24 hours and incubated at 37°C with 5% CO2 and 10% humidity. The floating cells from each condition were stored in conical tubes and the attached cells were washed with PBS (Corning) and removed by incubating them with 0.25% trypsin 2.21mM EDTA solution (Corning). The trypsin was diluted with 10ml DMEM supplemented medium and removed from the cells by centrifuging for 5 mins at 1000 RPM and aspirating the supernatant. The cells were resuspended in binding buffer (10 mM HEPES/ NaOH pH7.5 containing 0.14 M NaCl and 2.5mM CaCl2) and transferred to test tubes. Annexin V FITC Conjugate (1 µg/ml) (Sigma) and Propidium Iodide (1 µg/ml) (Sigma) were added to all tubes, including an untreated control, excluding a untreated, unstained control. The cells were incubated with the dyes for 10 minutes in the dark. Flow cytometry was done using a Becton Dickinson Fortessa SORP 14-color flow cytometer equipped with a 488nm excitation filter, a 515/20 nm emission filter and a 610/20 nm emission filter.
Results and discussion
Current receptor- ligand binding theory is hinged on the tenet that a given ligand will bind to a subset of accessible receptor conformations on the receptor's free energy landscape.Citation6 Depending on the nature of receptor conformations occupied, the number of the classic agonist versus antagonist conformations occupied, a “graded” or full receptor response will be rendered. WAY-169916 which is an ERα partial agonist, induces a graded response.Citation6 Ligand binding modifies the receptor by changing surface pockets on the receptor and thus effects coregulator selection. A dynamic ligand can bind to the receptor in multiple different orientations and a constrained ligand binds to the receptor in one orientation.Citation59 However, conformational dynamics of the receptor and ligand are not well understood. Cirillo et al using tandem affinity purification of tagged-ER followed by mass spectrometry, identified that most ER-protein interactions were ligand specific, 200 in the case of E2, 15 for raloxifene and 21 for fulvestrant.Citation60 Some examples of the ligand specific interactions generated are ER-fulverstrant's recruitment of KIAA1967 protein, which is known as deleted in breast cancer gene 1, and ER-tamoxifen's interaction with the α subunit of pyruvate dehydrogenase (PDHA1).Citation60 This indicates that the antiestrogens with “fixed-ring structures” that are currently used could influence only a sub-set of ER interactions and essentially makes the case for the discovery of new “flexible” ligands that could exploit most of the anti-estrogen specific proteins interactions.
We describe here a library of compounds with a morpholinoaniline scaffold, generated based on the ER LBD targeting pharmacophoric features from diethylstilbestrol (DES) with the potential to influence a wide range of ER-protein interactions. Appendages were added to the morpholinoaniline core structure to thoroughly explore the ER LBD and in hopes of attaining a group of ligands that could occupy a larger percentage of the receptor conformations available on the free energy landscape and render them inactive.
Substituted morpholinoaniline derivatives as potential ER modulators
In silico docking was performed with a library of 70 substituted morpholinoaniline derivatives termed “BR compounds” (). Four ER LBD X-ray crystal structures were used for the docking experiments: 3ERD (ERα agonist confirmation), 1ERR (ERα antagonist), 1QKM (ERβ partial agonist), and 1U3S (ERβ agonist). E2, 4OHT, raloxifene, and genistein were used as control ligands. Both autodock vina and sybyl were able to differentiate between the relative binding affinities of the controls to specific ER conformations. In addition both programs were able to successfully predict relative binding affinities (RBA) of selective estrogen receptor modulators (SERMs), 4OHT and raloxifene, which had higher docking scores when docked to 1ERR (antagonist conformation) and lower docking scores when docked to 3ERD, 1QKM and 1U3S (agonist or partial agonist conformations). Tamoxifen's autodock vina docking scores were as follows: −8.8 Kcal/mol (1ERR), −5.2(3ERD), −4.6(1QKM), −5.3(1U3S), with the highest predicted binding affinity for Tamoxifen-1ERR complex. Raloxifene's autodock vina docking scores were as follows:-10.6(1ERR), −6.8(3ERD), −5.6(1QKM), 0.6(1U3S). Tamoxifen's sybyl consensus docking scores were as follows: 10.33(1ERR), 0.02(3ERD), −10.63(1QKM), and 2.14(1U3S), again with the highest predicted binding affinity for Tamoxifen-1ERR complex. Raloxifene's sybyl docking scores were as follows: 10.99 (1ERR), −9.8(3ERD),-9.84(1QKM), −2.08(1U3S).
Figure 2. A heatmap of docking scores generated from BR Compounds mimetics. Autodock vina and Sybyl X were used to generate all docking scores displayed. The scoring gradient goes from black (low) to green (high docking score). The X-ray structures 3ERD, 1U3S, 1ERR and 1QKM structures represent ER in its agonist, agonist, antagonist and partial agonist conformations respectively. The binding poses of BR 10, 41, 46, and 51 indicate potential ER sub-type selectivity.
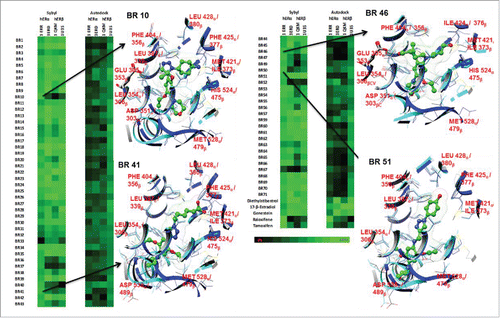
The agonists E2, diethylstilbestrol, and genistein predictably had higher docking scores in 3ERD, 1QKM, and 1U3S complexes, and lower docking scores in 1ERR in both Sybyl and Autodock Vina. E2, diethylstilbestrol, and genistein had the following autodock vina docking scores respectively: E2, −8.5 (1ERR), −7.9(3ERD), −9.3(1QKM), −10.8(1U3S); diethylstilbestrol, −7.1(1ERR), −8.9(3ERD), −6.5(1QKM), −9.8(1U3S); genistein, −7.4(1ERR), −9(3ERD), −8.1(1QKM), −9.6(1U3S). E2, diethylstilbestrol, and genistein had the following sybyl docking scores respectively: E2, 4.43 (1ERR), 6.32(3ERD), 6.44(1QKM), 7.24(1U3S); diethylstilbestrol, 6.13(1ERR), 7.89(3ERD), 8.93(1QKM), 6.02(1U3S); genistein, 6.13(1ERR), 7.4(3ERD), 7.32(1QKM), 6.3(1U3S). The predicted trend is similar to observed biological data (Kuiper, George, 1998). In addition overall docking results show that autodock vina scores result in a smaller range with the high and low scores equaling −10.8 and 0.6 respectively and sybyl docking scores fell in a larger range, with the high and low scores equaling 10.99 and −27.76. Due to a wider data spread, it was assumed that sybyl scores were able to better discriminate between small changes in binding interactions affording a wider range of docking scores. Of the 70 compounds in the library, 35 bound to the 1ERR with docking scores over the median binding score and 33 bound to 3ERD, 1QKM, 1U3S with docking scores over the median binding score. Thus the libraries of compounds produced include both potential agonists and antagonists.
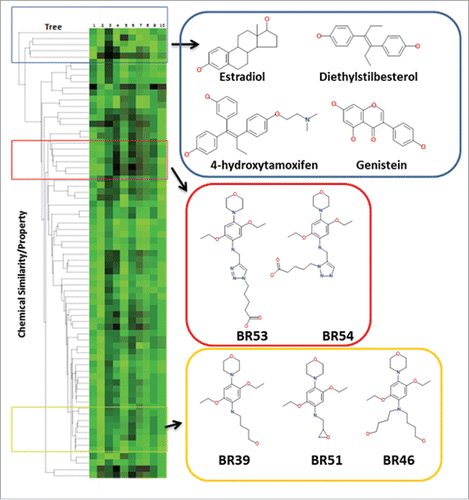
Of the 70 compounds, 11 are potential selective modulators targeting the ER antagonist conformation, as they were found to afford docking scores above the median docking score (used as a cut-off score) for 1ERR as compared with the other ER crystal structures. A dendrogram was generated in ChemMine tools and paired with the total docking scores from sybyl to determine the structural diversity of this compound library and prototype estrogenic ligands (). The dendrogram was generated with an all-against-all compound comparison and the distances between each compound was determined by the following equation 1-Tc, where Tc is the Tanimoto coefficient, in which the values range from 0 to 1. The dendrogram indicates that the BR Compounds are structurally distinct from the control ER ligands (distances > 0.65).
Figure 3. A heatmap of total binding scores and the chemical properties of each compound paired with a dendrogram. The dendrogram (ChemMine tools to compare the structure of each compound to the known ER modulators, and relate each compounds chemical structure to docking scores. Sybyl consensus binding score of each compound in complex with ER antagonist and agonist conformations is displayed in column 1 and 2 of the heatmap, respectively (rendered black (low) to green (high docking scores)). The chemical properties of each compound are displayed in column 3–10 (CLogP, compound area, polar surface area, polar volume, compound volume, molecular weight, H-bond aceptor, H-bond donor).
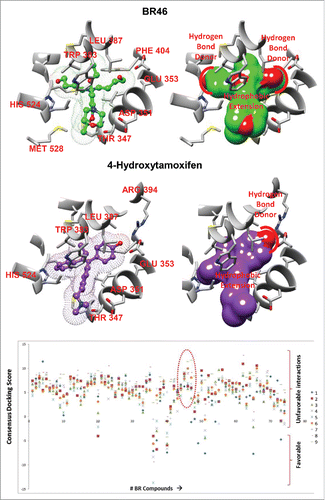
Only 4 out of the 70 compounds did not obey Lipinski's rule of 5. While the BR compounds were found to be structurally distinct, BR46, rendered a similar docking pose with respect to 4OHT in the ERα antagonist conformation, as both compounds make a hydrogen bond interaction with GLU 353 and have a hydrophobic core that precludes helix 12 from taking on the agonist spatial orientation ( and ). BR46 makes an additional hydrogen bond interaction with HIS 524. BR46 was one of the compounds in the BR library that had a high docking score in the 1ERR structure, but not the 3ERD structure. In fact there was approximately a 29 fold increase in the docking score of BR46 to 1ERR when compared with 3ERD indicating preference to antagonist conformation of ER.
Figure 4. Top docking pose of (A) BR46 and (B) 4-hydroxytamoxifen (4OHT). Key residues from the ligand binding pocket of the estrogen receptor highlighted in red. (C) Docking results of BR compounds in an ensemble of MD generated ER structures. A scatter plot of consensus docking scores obtained with the BR compounds were docked to an ensemble of MD generated structures ER agonist conformations(#1–9). A number of BR compounds including BR46, 47 (circled red) show less preference for ER agonist conformations.
BR compounds with N,N-dialkylated moieties consisting of different lengths of flexible carbon chains were designed to capture a larger pool of ER conformations. Therefore a more robust library of ER conformations was generated by subjecting the 3ERD crystal structure to molecular dynamic simulations in MOE (). The molecular dynamic simulations generated 9 lower energy conformations with variations in HIS 524. The BR compounds were then docked to the 9 MD generated ER structures. This experiment focused on determining how small changes in the agonist conformation of ER affected ligand binding. Eight of the 70 BR compounds were found to have docking scores less than the median docking score. These compounds were classified as potential ER antagonist. In addition 27 compounds were found to produce a docking score of less than the median docking score 2 or less ER MD generated structures. N,N-dialkylated morpholinoaniline derivaties, BR46 the compound that bound similar to OHT in antagonist conformations of ER, did not bind to any MD generated ER agonist structures was selected along with its structural homolog BR47 for further in vitro studies.
BR46 and BR47 alter cell viability in ER positive breast cancer cells
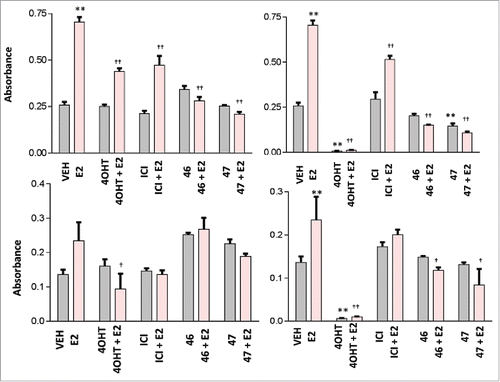
MCF7 and MDA-MB-231 cells were subjected to 0.5% DMSO (vehicle) or 0.05 to 50 µM of test compound treatments of 24 hours and the 3–4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium (MTT) assay was used to determine the number of viable cells. The ER negative cell line MDA-MB-231 was assayed as a negative control. BR46 and BR47 are proposed as ER targeting compounds, therefore it was hypothesized that BR46 and 47 would have no effect on MDA-MB-231 cell viability, in that this cell line lacks full length ER. After 24 hour treatments BR46 (0.5–50 µM) and BR47 (50 µΜ) did significantly reduce the number of viable ER positive MCF7 breast cancer cells but not the viability of ER negative MDA-MB-231 breast cancer cells (). Furthermore, to assess whether the compounds block E2 activity, MCF7 and MDA-MB-231 cells were subjected to 1 or 100 µM of test compounds and 4OHT treatments of 24 hours with and without 1nM E2. BR46 did significantly inhibit cell viability of ER positive MCF7 breast cancer cells at both the 1μM and the 100μM concentration ( & ). Also at the 100 uM concentration BR47 significantly reduced the number of viable cells in the absence of E2. BR46 and BR47 did not significantly affect the ER negative MDA-MB-231 breast cancer cell viability at 1 µM concentrations in the presence and absence of E2 (). However at 100 µΜ concentrations both compounds decreased cell viability of MDA-MB-231 cells ().
Figure 5. BR compounds effects on (A) MCF7 (ER positive) and (B) MDA-MB-231 (ER negative) breast cancer cell viability. MCF7 cells were treated with 0.5% DMSO (vehicle) or 0.05 to 50 uM test compounds for 24 hours at 37oC with 5% CO2 and 10% humidity. was used as vehicle. The cells were incubated with (1mM) thiazolyl bromide for 4 hours and the absorbance was captured at 570nm and 650nm using the Bio-Tek Synergy microplate reader. All assays were run in triplicates. Error analysis and statistics was performed using GraphPad Prism. Results represent the mean ± standard deviation, where *** represents p < 0.0001.
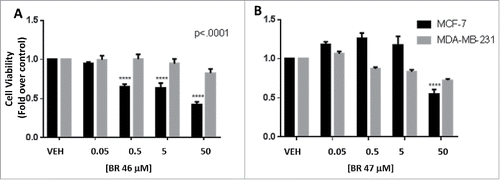
Figure 6. Effect of BR compounds on MCF7 breast cancer cell viability at 1 uM (A) and 100 uM (B) and on MDA-MB-231 cell line at 1 uM (C) and 100 uM (D). MCF7 cells were treated with 1 or 100 uM test compound or 4OHT, in the presence and absence of E2, for 24 hours at 37oC with 5% CO2 and 10% humidity. 0.5% DMSO was used as vehicle and 1nM E2 was used as a positive control. The cells were incubated with (1mM) thiazolyl bromide for 4 hours and the absorbance was captured at 570nm and 650nm using the Bio-Tek Synergy microplate reader. All assays were run in triplicates. Error analysis and statistics was performed using GraphPad Prism. Results represent the mean ± standard deviation, where * or † represents p < 0.05 and ** or †† represents p < 0.01. Two-way ANOVA analysis was performed with a Bonferroni multiple comparison posttest. The * was used to identify statistically significant data from the treatment vs. vehicle comparison. The † was used to identify statistically significant data from the treatment and E2 vs. E2 alone comparison.
BR46 and BR47 inhibit MCF7 breast cancer cell proliferation in the presence of estrogen
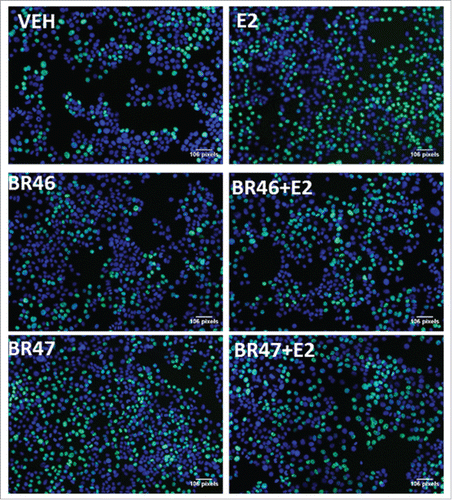
MCF7 breast cancer cells were subjected to 5 uM treatments of the test compounds (concentration selected based on cell viability data) in the presence or absence of E2 (1 nM) or 4OHT(5 uM). After 8 hours of treatment the compound effects were then resolved with a 5-ethynyl-2′-deoxyuridine (EdU) Click-iT HCS cell proliferation assay. Both BR46 and BR47 inhibited E2 (1nM) induced MCF7 cell proliferation (). BR46 and BR47 had no effects on MCF7 breast cancer cell proliferation in the presence of 4OHT. In addition, BR46 had no effects on MCF7 breast cancer cell proliferation in the absence of any other treatments. However, BR47 stimulated MCF7 breast cancer cell proliferation in the absence of other treatments. Representative figures from the EdU cell proliferation assay were captured with a Zeiss Axio Observer inverted microscope at 10X magnification with filters appropriate for Hoechst and FITC fluorescence (Alexa Fluor 488- ex495nm, em519nm; NuclearMask Blue stain- ex350nm, em461nm) to gauge the extent of EdU incorporation (). Both compounds did not alter cell viability in MDA-MB-231 cells (ER negative). shows significant increase in EdU post E2 treatment and subsequent reduction in EdU incorporation in BR46 and BR47 treatments in the presence of E2. BR46 alone did not induce a significant increase in cell proliferation however BR47 increased cell proliferation of MCF7 breast cancer cells when treated alone ().
Figure 7. BR compounds effects on MCF7 breast cancer cell proliferation. MCF7 cells were treated with 5uM of test compound for 8 hours at 37°C with 5% CO2 and 10% humidity. 0.1% DMSO was used as vehicle and 1nM E2 was used as a positive control. A Click-iT 5-ethylnyl-2′-deoxyuridine (EDU) Alexa Fluor 488 based HCS assay was then performed in the 96 well format. The fluorescence intensity was captured using 495nm excitation and 519nm emission filters with the Bio-Tek Synergy plate reader. All assays were run in triplicates and at least 3 independent experiments. Error analysis and statistics was performed using GraphPad Prism. Results represent the mean ± standard deviation, where * or † represents p < 0.05 and ** or †† represents p < 0.01. Two-way ANOVA analysis was performed with a Bonferroni multiple comparison posttest. The * was used to identify statistically significant data from the treatment vs. vehicle comparison. The † was used to identify statistically significant data from the treatment and E2 vs. E2 alone comparison.
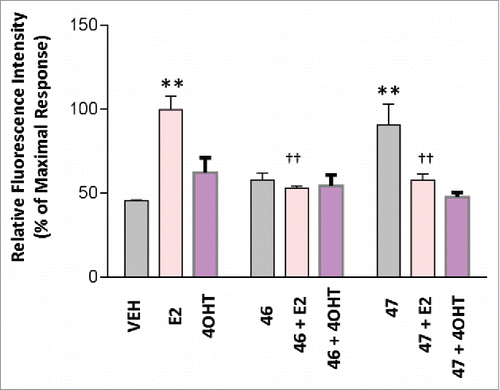
Figure 8. Representative images of the EdU cell proliferation assay done after MCF7 cells were subjected to BR compound treatments. The images were captured with a Zeiss Axio Observer inverted microscope at 10X magnification with filters appropriate for Hoechst and FITC fluorescence (Alexa Fluor 488- ex495nm, em519nm; NuclearMask Blue stain- ex350nm, em461nm). The total DNA content was stained with a NuclearMask Blue stain (blue), and EdU was visualized after conjugation with Alexa Fluor 488 azide (green).
BR compounds did not induce apoptosis in breast cancer cells
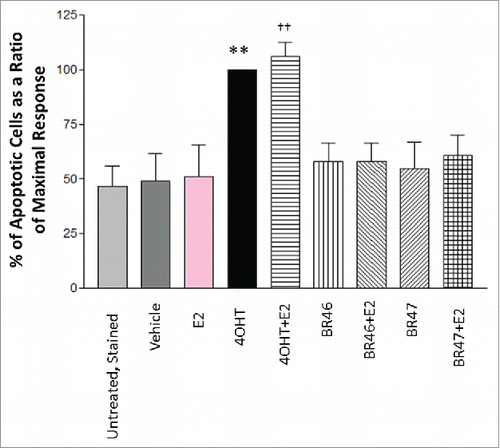
BR compounds, purposefully designed to act as targeted therapies and inhibit aberrant cell proliferation, were found to attenuate E2 induced MCF7 breast cancer cell proliferation and cell viability, it is important to assess whether they are cytostatic or cytotoxic. To determine if the test compounds induced MCF7 and MDA-MB-231 (negative control) breast cancer cell apoptosis, the cells were subjected to 5 uM test compound treatments of 16 hours and the number of cells undergoing apoptosis was evaluated with Annexin V and propidium iodide staining using Becton Dickinson Fortessa SORP 14-color flow cytometer equipped with a 488nm excitation filter, a 515/20 nm emission filter and a 610/20 nm emission filter. None of the test compounds induced apoptosis in either MCF7 cells () or MDA-MB-231 (not shown).
Figure 9. BR compounds effects on MCF7 breast cancer cell apoptosis. MCF7 cells were treated with 5 uM BR compounds and/or 1nM E2 for 16 hours and incubated at 37oC with 5% CO2 and 10% humidity. 0.1% DMSO was used as vehicle and 1 uM 4OHT was used as a positive control. Cell death was gauged by staining with Annexin V FITC Conjugate (1ug/ml) and Propidium Iodide (1ug/ml). Flow cytometry was done using a Becton Dickinson Fortessa SORP 14-color flow cytometer equipped with a 488nm excitation filter, a 515/20nm emission filter and a 610/20nm emission filter. The assay was run in 3 independent experiments. Results represent the mean ± standard deviation, where * or † represents p < 0.05 and ** or †† represents p < 0.01. Two-way ANOVA analysis was performed with a Bonferroni multiple comparison posttest. The * was used to identify statistically significant data from the treatment vs. vehicle comparison. The † was used to identify statistically significant data from the treatment and E2 vs. E2 alone comparison.
Conclusions
A morpholinoaniline scaffold was chosen as the ideal template to derivatize and generate a library of ligands (BR compounds) targeting ER LBD. The hydroxyl groups make hydrogen bond interactions with ER residues HIS 524, GLU 353 and ARG 394. This pharmacophore is consistent in both ER agonist and antagonist ligands. In silico analysis suggest that the morpholinoaniline core could also accommodate posttranslational modifications, specifically the phosphorylation of TYR 537 and THR 347, which are implicated in tamoxifen resistance.Citation16 Compounds were designed by adding flexible moieties to the morpholinoaniline scaffold at the 2, 4 and 5 position of the aromatic aniline ring. The anilinic nitrogen was either N-monoalkylated or N,N-dialkylated. The appendages added to the anilinic nitrogen (2- position of the scaffold) contained a hydroxy group at the end to provide the pharmacophoric features needed to form hydrogen bonding interactions with the estrogen receptor binding pocket. It was important to incorporate flexibility in the compound design in order for the compounds to bind to as many antagonist conformations of the ER landscape as possible. In addition, it has been shown that compounds that promote ER heterodimerization lead to inhibition of breast cancer cell proliferation and ER transactivation.Citation61 Compounds with smaller spacer connecting the pharmacophoric units such as actinopolymorphol A induce ER heterodimerization.Citation62 This suggested that the ER LBD may constrict when forming the heterodimer. Flexible compounds would be able to adapt to a constricted ER LBD and potentially induce ER heterodimers in addition to ER antagonist conformations. Therefore, for the first time, a library of flexible compounds that can bind to multiple ER conformations and render them inactive either through promoting ER protein-protein interactions with co-repressors or by reorienting helix 12 thereby blocking co-activator recruitment.
In silico docking studies showed that 35 out of the 70 morpholinoaniline derivatives from the BR compound library bound to the antagonist conformation of ER and 33 of the designed compounds bound to the agonist and partial agonist conformations with docking scores above the median binding scores of reference ligands. Thus the library of compounds generated includes both potential agonist and antagonist of ER. Cheminformatics analysis showed the compounds designed are structurally distinct from the known ER LBD targeting compounds. Since the compounds were designed to capture multiple antagonist conformations, it was important to determine if the compounds would bind to agonist conformation as well. Only 8 of the 70 BR compounds bound with less affinity (below the docking threshold) to the ensemble MD generated ER agonist conformations.
Lead compounds BR46 and BR47 were tested to determine their effects on breast cancer cell proliferation, cell viability and cell death. BR46 and BR47 inhibited E2 induced cell proliferation and cell viability in ER positive MCF7 breast cancer cells, but had no effects on ER negative MDA-MB-231 breast cancer cells, suggesting that BR46 and BR47 act through an ER dependent mechanism.
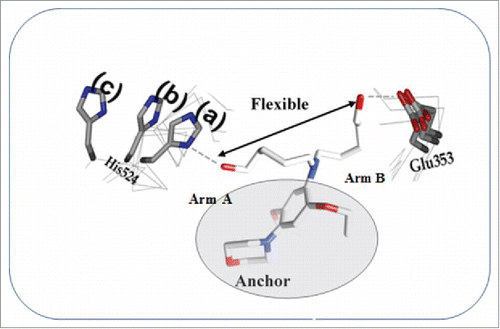
A model of the BR46 compound interaction with the multiple ER conformations is shown in . It would be interesting to determine if any of the BR compounds recruit several corepressors by presenting viable motifs at the binding interface as needed. Furthermore, this could provide a better understanding of BR compounds' mechanism of action. We are currently preforming studies directed at rigidification of Arm B, as only HIS 524 of the ER LBD takes on different conformations when bound to various ER/antiestrogen/co-repressor complexes. In addition, BR46 should be tested in vivo to determine its effects on breast cancer tumor growth. We believe the structural clues generated in this study are useful in generating a new breed of antiestrogens targeting multiple ER conformations.
Figure 10. The lead compound BR 46 capturing an ensemble of ER conformations.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The authors declare that this work was supported in part by the Jacobs School of Medicine and Biomedical Science funds and USPHS P20 MD002725 to RVR.
References
- Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol Endocrinol 2005; 19:833-42; PMID:15695368; https://doi.org/10.1210/me.2004-0486
- Levin ER. Integration of the extranuclear and nuclear Actions of Estrogen. Mol Endocrinol 2005; 19:1951-9; PMID:15705661; https://doi.org/10.1210/me.2004-0390
- Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, Giguère V. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor β. Mol Endocrinol 1997; 11:353-65; PMID:9058381; https://doi.org/10.1210/mend.11.3.9902
- Enmark E, Pelto-Huikko M, Grandien K, Lagercrantz S, Lagercrantz J, Fried G, Nordenskjöld M, Gustafsson JA. Human Estrogen Receptor β-Gene Structure, Chromosomal Localization, and Expression Pattern. J Clin Endocrinol Metab 1997; 82:4258-65; PMID:9398750; https://doi.org/10.1210/jcem.82.12.4470
- Ponglikitmongkol M, Green S, Chambon P. Genomic organization of the human oestrogen receptor gene. EMBO J 1988; 7:3385-8; PMID:3145193
- Bruning JB, Parent AA, Gil G, Zhao M, Nowak J, Pace MC, Smith CL, Afonine PV, Adams PD, Katzenellenbogen JA, et al. Coupling of receptor conformation and ligand orientation determine graded activity. Nat Chem Biol 2010; 6:837-43; PMID:20924370; https://doi.org/10.1038/nchembio.451
- Beato M, Sanchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev 1996; 17:587-609; PMID:8969970; https://doi.org/10.1210/edrv-17-6-587
- Cavailles V, Dauvois S, Danielian PS, Parker MG. Interaction of protein with transcriptionally active estrogen receptors. Proc Natl Acad Sci U S A 1994; 91:10009-13; PMID:7937828; https://doi.org/10.1073/pnas.91.21.10009
- L'Horset F, Dauvois S, Heery DM, Cavailles V, Parker MG. RIP-140 interacts with multiple nuclear receptors by means of two distinct sites. Mol Cell Biol 1996; 16:6029-36; PMID:8887632; https://doi.org/10.1128/MCB.16.11.6029
- vom Baur E, Zechel C, Heery DM, Heine MJ, Garnier JM, Vivat V, Le Douarin B, Gronemeyer H, Chambon P, Losson R. Differential ligand-dependent interactions between AF-2 activating domain of nuclear receptors and the putative transcriptional intermediary factors mSUG1 and TIF1. EMBO J 1996; 15:110-24; PMID:8598193
- Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S. Ligand-,cell-,and estrogen receptor subtype (alpha/beta)-dependent activation of GC-rich (Sp1) promoter elements. J Biol Chem 2000; 275:5379-87; PMID:10681512; https://doi.org/10.1074/jbc.275.8.5379
- Peters GA, Khan SA. Estrogen receptor domains E and F role in dimerization and interaction with coactivator RIP-140. Mol Endocrinol 1999; 13:286-96; PMID:9973258; https://doi.org/10.1210/mend.13.2.0244
- Hall JM, McDonnell DP, Korach KS. Allosteric regulation of estrogen receptor structure, function, and coactivator recruitment by different estrogen response elements. Mol Endocrinol 2002; 16:469-86; PMID:11875105; https://doi.org/10.1210/mend.16.3.0814
- O'Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol Endocrinol 2004; 18:1859-4875; PMID:15031323; https://doi.org/10.1210/me.2003-0044
- Wronski TJ, Clintron M, Doherty SL, Dann LM. Estrogen treatment prevents osteopenia and depresses bone turnover in ovariectomized rats. Endocrinology 1988; 123:681-6; PMID:3396505; https://doi.org/10.1210/endo-123-2-681
- Riggins R, Schrecengost R, Guerrero M, Bouton A. Pathways to tamoxifen resistance. Cancer Lett 2007; 256:1-24; PMID:17475399; https://doi.org/10.1016/j.canlet.2007.03.016
- Lerner LJ, Holthaus JF, Thompson CR. A non-steroidal estrogen antagonist 1-(p-2-diethylaminoethoxyphenyl)-1-phenyl-2-p-methoxyphenylethanol. Endocrinology 1958; 63:295-318; PMID:13574085; https://doi.org/10.1210/endo-63-3-295
- Howell S, Johnston SRD, Howell A. The use of selective estrogen receptor modulators and selective estrogen receptor down-regulators in breast cancer. Best Pract Res Clin Endocrinol Metab 2004; 18:47-66; PMID:14687597; https://doi.org/10.1016/j.beem.2003.08.002
- Jordan VC. Tamoxifen: A most unlikely pioneering medicine. Nat Rev Drug Discovery 2003; 2:205-13; PMID:12612646; https://doi.org/10.1038/nrd1031
- Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer 2004; 11:643-58; PMID:15613444; https://doi.org/10.1677/erc.1.00776
- Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, Abraham M, Medeiros Alencar VH, Badran A, Bonfill X, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013; 381:805-15; PMID:23219286; https://doi.org/10.1016/S0140-6736(12)61963-1
- Kangas L, Nieminen AL, Blanco G. A new triphenylethylene compound, Fc-1157a. II. Antitumor effects. Cancer Chemother Pharmacol 1986; 17:109-13; PMID:2941177; https://doi.org/10.1007/BF00306737
- Chander SK, McCague R, Luqmani Y, Newton C, Dowsett M, Jarman M, Coombes RC. Pyrrolidino-4-iodatamifen and 4-iodotamoxifen, new analogues of the antiestrogen tamoxifen for the treatment of breast cancer. Cancer Res 1991; 51:5851-8; PMID:1933854
- Loser R, Seibel K, Eppenberger U. No loss of estrogen or anti-estrogenic activity after demethylation of droloxifene (3-OH-tamoxifen). Int J Cancer 1985; 36:701-3; PMID:4066073; https://doi.org/10.1002/ijc.2910360614
- Toko T, Sugimoto Y, Matsuo K. TAT-59, a new triphenylethylene derivative with antitumor activity against hormone-dependent tumours. Eur J Cancer Clin Oncol 1990; 26:397-404; PMID:2141500; https://doi.org/10.1016/0277-5379(90)90241-K
- Toko T, Shibata J, Nukatsuka M. Antiestrogenic activity of DP-TAT-59, an active metabolite of TAT-59 against human breast cancer. Cancer Chemother Pharmacol 1997; 39:390-8; PMID:9054952; https://doi.org/10.1007/s002800050589
- Palkowitz AD, Glasebrook AL, Thrasher KJ. Discovery and synthesis of [6-hydroxy-3-[4-[2-(1-piperidinly)ethoxy]phenoxy]-2-(4-hydroxyphenyl)]b enzo[b]thiophene: A novel, highly potent, selective estrogen receptor modulator. J Med Chem 1997; 40:1407-16; PMID:9154963; https://doi.org/10.1021/jm970167b
- Clemens JA, Bennett DR, Black LJ. Effects of a new antiestrogen, keoxifene (LY 156758), on growth of carcinogen-induced mammary tumours and on LH and prolactin levels. Life Sci 1983; 32:2869-75; PMID:6406781; https://doi.org/10.1016/0024-3205(83)90323-5
- Gauthier S, Caron B, Cloutier J. (S)-(+)-4-[7-(2,2-dimethyl-1-oxopropoxy)-4-methyl-2[4-[2-(1-piperidinyl)-ethoxy]phenyl]-SH-1-benzopyran-3-yl]-phenyl 2, 2-dimethylpropanoate 9EM-800): A highly potent, specific, and orally active nonsteroidal antiestrogen. J Med Chem 1997; 40:2117-22; PMID:9216828; https://doi.org/10.1021/jm970095o
- Jones CD, Suarez T, Massey EH, Black LJ, Tinsley FC. Synthesis and antiestrogenic activity of [3,4-dihydro-2-(4-methoxyphenyl)-1-naphthalenyl]methanone, methanesulfonic acid salt. J Med Chem 1979; 22:962-6; PMID:490541; https://doi.org/10.1021/jm00194a015
- Coopman P, Garcia M, Brunner N. Anti-proliferative and anti-estrogenic effects of ICI 164,384 and ICI 182,780 in 4-OH-tamoxifene-resistant human breast cancer cells. Int J Cancer 1994; 56:295-300; PMID:8314314; https://doi.org/10.1002/ijc.2910560225
- Brunner N, Frandsen TL, Holst-Hansen C. MCF7/LCC2: A 4-hydroxytamoxifen resistant human breast cancer variant that retains sensitivity to the steroidal antiestrogen ICI 182,780. Cancer Res 1993; 53:3229-32; PMID:8324732
- Hu XF, Veroni M, De Luise M. Circumvention of tamoxifen resistance by the pure anti-estrogen ICI 182,780. Int J Cancer 1993; 55:873-6; PMID:8244585; https://doi.org/10.1002/ijc.2910550529
- Brunner N, Boysen B, Jirus S. MCF7/LCC9: An antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res 1997; 57:3486-93; PMID:9270017
- Osborne CK, Coronado-Heinsohn EB, Hilsenbeck SG. Comparison of the effects of a pure antiestrogen with those of tamoxifen in a model of human breast cancer. J Natl Cancer Inst 1995; 87:746-50; PMID:7563152; https://doi.org/10.1093/jnci/87.10.746
- Wakeling AE, Dukes M, Bowler JA. A potent specific pure antiestrogen with clinical potential. Cancer Res 1991; 51:3867-73; PMID:1855205
- Parker MG. Action of ‘pure’ antiestrogens in inhibiting estrogen receptor action. Breast Cancer Res Treat 1993; 26:131-7; PMID:8219250; https://doi.org/10.1007/BF00689686
- Kuiper G, Gustafsson J-Å. The novel estrogen receptor-beta subtype: Potential role in the cell- and promoter-specific actions of estrogens and anti-estrogens. FEBS Lett 1997; 410:87-90; PMID:9247129; https://doi.org/10.1016/S0014-5793(97)00413-4
- Katzenellenbogen BS, Korach KS. A new actor in the estrogen receptor drama- enter ER-beta. Endocrinology 1997; 138:861-2; PMID:9048583; https://doi.org/10.1210/endo.138.3.5080
- Mosselman S, Polman J, Dijkema R. ER beta: Identification and characterization of a novel human estrogen receptor. FEBS Lett 1996; 392:49-53; PMID:8769313; https://doi.org/10.1016/0014-5793(96)00782-X
- Webb P, Nguyen P, Kushner PJ. Differential SERM effects on corepressor binding dictate ER alpha activity in vivo. J Biol Chem 2003; 278:6912-20; PMID:12482846; https://doi.org/10.1074/jbc.M208501200
- Wijayaratne AL, McDonnell DP. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonist, and selective estrogen receptor modulators. J Biol Chem 2001; 276:35684-92; PMID:11473106; https://doi.org/10.1074/jbc.M101097200
- Hoffman J, Bohlmann R, Heinrich N. Second generation of estrogen receptor destabilising compounds-pharmacological profile in antiestrogen-sensitive or -resistant breast cancer. Breast Cancer Res Treat 2002; 76:27; PMID:12408373
- Kieser KJ, Kim DW, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Characterization of the pharmacophore properties of novel selective estrogen receptor downregulators (SERDs). J Med Chem 2010; 53:3320-9; PMID:20334372; https://doi.org/10.1021/jm100047k
- McDonnell DP, Clemm DL, Hermann T, Goldmann ME, Pike JW. Analysis of estrogen receptor function in vitro reveals three distinct classes of antiestrogens. Mol Endocrinol 1995; 9:659-69; PMID:8592512; https://doi.org/10.1210/mend.9.6.8592512
- Wijayaratne AL, Nagel SC, Paige LA, Christensen DJ, Norris JD, Fowlkes DM, McDonnell DP. Comparative analyses of mechanistic differences among antiestrogens. Endocrinology 1999; 140:5828-40; PMID:10579349; https://doi.org/10.1210/endo.140.12.7164
- MacGregor-Shafer JI, Liu H, Bentrem D, Zapf J, Jordan VC. Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex by substituting glycine for aspartate at amino acid 351. Cancer Res 2000; 60:5097-105; PMID:11016635
- Norris JD, Paige LA, Christensen DJ, Chang CY, Huacani MR, Fan D, Hamilton PT, Fowlkes DM, McDonnell DP. Peptide antagonists of the human estrogen receptor. Science 1999; 285:744-6; PMID:10426998; https://doi.org/10.1126/science.285.5428.744
- Paige LA, Christensen DJ, Gronemeyer H, Norris JD, Gottlin EB, Padilola KM, Chang CY, Ballas LM, Hamilton PT, McDonnell DP, et al. Estrogen receptor (ER) modulators each induce distinct conformational changes in ER alpha and ER Beta. Proc Natl Acad Sci U S A 1999; 96:3999-4004; PMID:10097152; https://doi.org/10.1073/pnas.96.7.3999
- Levenson AS, MacGregor-Shafer JI, Bentrem D, Pease KM, Jordan VC. Control of the estrogen-like actions of tamoxifen-estrogen receptor complex by the surface amino acid at position 251. J Steroid Biochem Mol Biol 2001; 76:61-70; PMID:11384864; https://doi.org/10.1016/S0960-0760(00)00143-6
- Wolf DM, Jordan VC. Characterization of tamoxifen stimulated MCF-7 tumor variants growth in athymic mice. Breast Cancer Res Treat 1994; 31:117-27; PMID:7981452; https://doi.org/10.1007/BF00689682
- Wolf DM, Jordan VC. The estrogen receptor from a tamoxifen stimulated MCF-7 tumor variant contains a point mutation in the ligand binding domain. Breast Cancer Res Treat 1994; 31:129-38; PMID:7981453; https://doi.org/10.1007/BF00689683
- Levenson AS, Tonetti DA, Jordan VC. The oestrogen-like effect of 4-hydroxytamoxifen on induction of transforming growth factor alpha mRNA in MDA-MB-231 breast cancer cells stably expressing the oestrogen receptor. Br J Cancer 1998; 77:1812-9; PMID:9667651; https://doi.org/10.1038/bjc.1998.301
- Wen J, Li R, Lu Y, Shupnik M. Decreased BRCA1 confers tamoxifen resistance in breast cancer cells by altering estrogen receptor-coregulator interactions. Oncogene 2009; 28:575-86; PMID:18997820; https://doi.org/10.1038/onc.2008.405
- Backman TW, Cao Y, Girke T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res 2011; 39:W486-91; PMID:21576229; https://doi.org/10.1093/nar/gkr320
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res 2000; 28:235-42; PMID:10592235; https://doi.org/10.1093/nar/28.1.235
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res 2017; 45:158-69; PMID:27899622; https://doi.org/10.1093/nar/gkw1099
- Chakraborty S, Asare BK, Biswas PK, Rajnarayanan RV. Designer interface peptide grafts target estrogen receptor alpha dimerization. Biochem Biophys Res Commun 2016; 478:116-22; PMID:27462021; https://doi.org/10.1016/j.bbrc.2016.07.083
- Srinivasan S, Nwachukwu JC, Parent AA, Cavett V, Nowak J, Hughes TS, Kojetin DJ, Katzenellenbogen JA, Nettles KW. Ligand-binding dynamics rewire cellular signaling via estrogen receptor-alpha. Nat Chem Biol 2013; 9:326-32; PMID:23524984; https://doi.org/10.1038/nchembio.1214
- Cirillo F, Nassa G, Tarallo R, Stellato C, Filippo MRD, Ambrosino C, Baumann M, Nyman TA, Weisz A. Molecular mechanisms of selective estrogen receptor modulator activity in human breast cancer cells: Identification of novel nuclear cofactors of antiestrogen–ERα complexes by interaction proteomics. J Proteome Res 2013; 12:421-31; PMID:23170835; https://doi.org/10.1021/pr300753u
- Powell E, Shanle E, Brinkman A, Li J, Keles S, Wisinski K, Huang W, Xu W. Identification of estrogen receptor dimer selective ligands reveals growth-inhibitory effects on cells that co-express ER alpha and ER beta. PLoS ONE 2012; 7:e30993; PMID:22347418; https://doi.org/10.1371/journal.pone.0030993
- Powell E, Huang SX, Xu Y, Rajski SR, Wang Y, Peters N, Guo S, Xu HE, Hoffmann FM, Shen B, et al. Identification and characterization of a novel estrogenic ligand actinopolymorphol a. Biochem Pharmacol 2010; 80:1221-9; PMID:20599778; https://doi.org/10.1016/j.bcp.2010.06.030