
ABSTRACT
P53 tumor suppressor gene mutations occur in the majority of human cancers and contribute to tumor development, progression and therapy resistance. Direct functional restoration of p53 as a transcription factor has been difficult to achieve in the clinic. We performed a functional screen using a bioluminescence-based transcriptional read-out to identify small molecules that restore the p53 pathway in mutant p53-bearing cancer cells. We identified CB002, as a candidate that restores p53 function in mutant p53-expressing colorectal cancer cells and without toxicity to normal human fibroblasts. Cells exposed to CB002 show increased expression of endogenous p53 target genes NOXA, DR5, and p21 and cell death which occurs by 16 hours, as measured by cleaved caspases or PARP. Stable knockdown of NOXA completely abrogates PARP cleavage and reduces sub-G1 content, implicating NOXA as the key mediator of cell death induction by CB002. Moreover, CB002 decreases the stability of mutant p53 in RXF393 cancer cells and an exogenously expressed R175H p53 mutant in HCT116 p53-null cells. R175H p53 expression was rescued by addition of proteasome inhibitor MG132 to CB002, suggesting a role for ubiquitin-mediated degradation of the mutant protein. In summary, CB002, a p53 pathway-restoring compound that targets mutant p53 for degradation and induces tumor cell death through NOXA, may be further developed as a cancer therapeutic.
Introduction
The TP53 gene encodes the tumor suppressor protein p53, known as “the guardian of the genome,” which ensures the fidelity of DNA replication and controls cell division, thereby preventing the formation and abnormal growth of cancerous cells. p53 becomes stimulated upon genotoxic and other cellular stress signals including DNA damage, loss of cell adhesion, spindle damage, oncogene activation, nutrient deprivation, ribonucleotide depletion, and hypoxia.Citation1,Citation2 Ultimately, such stresses lead to p53-mediated transcriptional activation of genes involved in DNA repair, cell cycle arrest, cellular senescence, and apoptosis. One of the most well studied outcomes of p53 has been apoptosis, owing to p53's irreversible capacity to induce programmed cell death. Among established p53 targets that participate in apoptosis are NOXA, PUMA, DR5, and Bax.
TP53 is mutated in more than 50% of all human cancers, and has been a pivotal cancer target for drug development. TP53 mutation is a poor prognostic marker in various types of cancer. Unlike other tumor suppressors, missense mutations are the most common in TP53 and can result in the expression of a stable mutated p53 protein.Citation3 TP53 mutations can result in loss of function (LOF), a dominant-negative phenotype, or gain-of-function (GOF) activity for the encoded mutant protein. Studies have shown in vitro and in vivo that introduction of certain types of p53 mutants in a p53-null background results in new phenotypes where tumor cells are more proliferative, invasive, resistant to therapy, or more metastatic.Citation4,Citation5
In addition to mutant p53 acting in a dominant-negative fashion toward wild-type p53, mutant p53 has been shown to inhibit p53 family proteins p73 and p63. Consequently, p73 and p63 become incapable of exerting their tumor suppressive functions. p73 and p63 are transcription factors that share significant structural homology with p53. Similar to p53, p73 and p63 control the expression of genes involved in cell cycle arrest and apoptosis. It has been shown that p73 and p63 can functionally replace p53.Citation6 Unlike p53, however, they are very rarely mutated in cancer. Therefore, restoration of the p53 pathway through its family members represents an attractive therapeutic approach.
Despite numerous efforts to identify small molecule compounds for mutant p53-targeted therapy, to date there is no approved drug that restores a functional p53 pathway in cancer cells with mutant p53. Given that TP53 is the most commonly mutated tumor suppressor, it is an attractive therapeutic strategy to identify such small molecules. With our current knowledge that p53 family members p73 and p63 can perform similar anti-tumor effects, our group and others have identified small molecules that restore the p53 pathway through the activation of p73. Using a luciferase-based p53-reporter, our group has previously identified several compounds that restore the p53 pathway including prodigiosin and NSC59984.Citation7-9 We reported that these compounds up-regulate p73 although the downstream mechanisms of action are believed to be different, and other regulatory activities of the molecules may be important. Furthermore, we believe that mutant p53 protein degradation is necessary for optimal p73-mediated p53 pathway restoration. These findings support the pursuit of therapeutic strategies that target mutant p53 for degradation.
P53-targeted therapy is challenging because direct functional restoration of p53 activity as a DNA-binding transcription factor has been difficult to achieve using approaches whose goal is to modify p53 protein structure. We have taken a different approach by investigating small molecules that functionally restore the p53-signaling pathway instead of requiring direct p53 protein binding. Our hypothesis is that adequate p53 restoration in cancer cells carrying mutated p53 may involve the removal or inactivation of mutant p53 protein and activation of p53 family members p73 and p63. Therefore, we screened for small molecules that functionally stimulated p53-dependent transcriptional activation and elicited a p53-like tumor suppression while simultaneously degrading mutant p53. We identified a novel small molecule named CB002 as a candidate for the restoration of the p53 pathway in p53-null, wild-type, or mutant p53-expressing colorectal cancer (CRC) cells. In this study, we characterized CB002 with regard to its activity as a p53 pathway-restoring small molecule. In addition, we investigated how CB002 mediates mutant p53 degradation and how it mediates tumor cell death.
Results
Small molecule CB002 restores p53-dependent transcriptional reporter activity
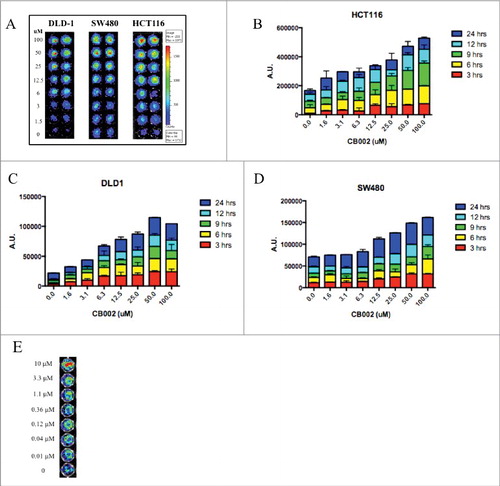
To identify small molecules that could restore the p53 signaling pathway, we screened 50,000 small molecules from the Chembridge Library using a firefly luciferase human p53 reporter assay system. SW480 colorectal cancer cells that stably express the human p53 reporter were treated with compounds at various concentrations from 0–100 μM for 2 and 24 hrs. This initial screen identified CB002 (ID 7745998, IUPAC name: 8-anilino-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione) as a small molecule capable of activating the luciferase reporter in a dose-dependent manner. To further validate the effects of CB002, we expanded the screening by testing effects on p53-dependent reporter activity in DLD-1, HCT116 (p53+/+), and HCT116 (p53−/−) CRC cell lines. In all cell lines tested, the activity of the reporter was induced in a dose-dependent manner at 3 and 24 hrs (). These results document that CB002 restores the p53-dependent transcriptional activity of a reporter gene in a panel of CRC cell lines.
Figure 1. CB002 activates luciferase-based p53-reporter activity in 3 different human colorectal cancer cell lines in a dose-dependent manner. A. Representative images of luciferase-based p53-reporter activity assays are shown at 3 hrs. B-D. Quantification of the luciferase-based p53-reporter activity assays in 3 different cell lines incubated from 3–24 hrs with CB002. E. Representative images of the p53-reporter activity assays are shown at 2 hrs for HCT116 p53-null cells. Three replicates were done for each concentration of CB002 as indicated in the figure panels.
A favorable therapeutic index is observed with CB002 using human tumor and normal cell lines
To evaluate the potential of CB002 as an anti-cancer agent, we first determined its therapeutic index by treating cancer (DLD-1, SW480, and RXF393) and normal (WI38 and MRC5) cell lines with concentrations ranging from 0 - 500 μM and assessing cell viability by the CellTiter-Glo® luminescence assay (). IC50 values were determined using GraphPad analyses and are listed in . CB002 has a significant therapeutic index among the cells tested. Normal cell lines have an IC50 value of approximately 650 μM while in the panel of cancer cell lines tested IC50 values ranged from 96 μM - 400 μM. SW480 was observed to be the most sensitive cell line, followed by DLD-1, and RXF393 is the least sensitive.
Figure 2. CB002 has a favorable therapeutic index. Cancer (DLD-1, SW480, and RXF393) and normal (WI38 and MRC5) cell lines were treated with CB002 with concentrations ranging from 0 - 500 μM for a period of 72 hrs, at this point cell viability was measured using the CellTiter-Glo® luminescence assay.
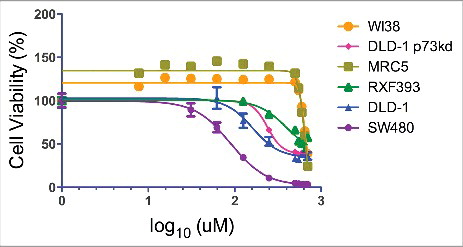
Table 1. CB002 IC50 values determined using GraphPad analyses for the cell lines tested in Figure 2.
Small molecule CB002 qualitatively decreases colony formation by colorectal cancer cell lines
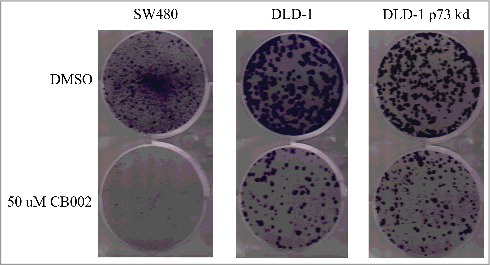
Colony formation assays are widely used to determine the ability of a single cell to proliferate and form a colony in culture. This method offers the advantage of elucidating the sensitivity of cells toward cytotoxic agents in a long-term assay that may mimic the response seen in mouse models. To determine the effect of CB002 on long-term cancer cell proliferation, we performed a colony formation assay in a panel of CRC cell lines. CB002 was observed to significantly decreases colony formation in SW480, DLD-1, and p73 DLD-1 stable knockdown cells (). These results suggest that CB002 has cytotoxic effects against human cancer cells in a long-term assay and that the effects may not depend on p73.
Figure 3. CB002 qualitatively decreases colony formation in SW480, DLD-1, and DLD-1 stable p73 knockdown colorectal cancer cell lines. Cells were treated with 50 μM CB002 for a period of 24 hrs, at which point media was replaced with complete media for a period of 15 d. Images are from one of 3 replicates.
CB002 induces apoptotic cell death of tumor cells
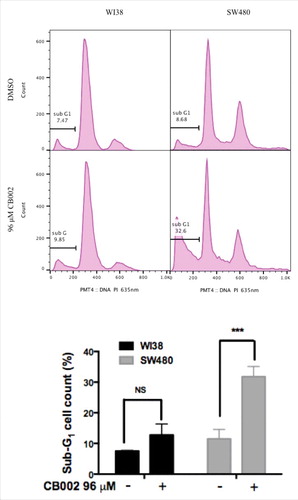
To determine whether CB002 promotes cancer cell death through apoptosis, we treated SW480 cells and subjected the cells to cell cycle profile analyses using propidium iodide staining. SW480 DMSO vehicle control sub-G1 population was 8.7% whereas treatment with 96 μM CB002 increased the sub-G1 population to 32.6% ( right panels). This 4-fold increase in sub-G1 content upon CB002 treatment indicates an augmentation in apoptotic cells. Furthermore, 96 μM CB002 treated WI38 normal cells showed a sub-G1 population of 9.8% as compared with the DMSO control where the sub-G1 content was 7.6% ( left panel). This increase in sub-G1 population in WI38 cells was determined not to be statistically significant ( bottom graph). Thus, CB002 induces cell death through apoptosis specifically in cancer cell lines but not in normal cells.
Figure 4. CB002 treatment of 48 hrs increases apoptotic cells as indicated by the sub-G1 content in SW480 cancer cells but not in normal WI38 cells. Two-way ANOVA statistical analysis, p ≤ 0.001 against DMSO vehicle control. Three replicates were performed, and a representative histogram is shown.
CB002 induces the expression of endogenous p53 target genes in mutant p53-expressing tumor cells and apoptotic cell death
To further investigate the potential of CB002 as a p53 pathway-restoring compound, we treated various cancer cell lines and probed for expression of endogenous p53 target genes, as well as for markers of apoptotic cell death. SW480 cells were treated with DMSO, 12, 25, and 50 μM CB002 as concentrations below the IC50 value. As shown in , CB002 was found to increase the expression of proteins involved in p53-dependent cell cycle arrest and apoptosis, including p21 and DR5 (, compare lane 1 to 2, 3, and 4). NOXA, a pro-apoptotic protein, was found to increase by CB002 concentrations below the IC50 (50 μM) and at the IC50 (96 μM) in SW480 cells (, compare lane 1 to 2, and 3). Altogether, these data demonstrate CB002-mediated induction of the p53 pathway in mutant p53 harboring cancer cells.
Figure 5. CB002 induces expression of p53 target genes independently of p73 and NOXA is required for CB002-mediated apoptosis. A-(C)Whole cell lysates were subjected to western blot analysis A. p73 was knocked-down by siRNA in SW480 cells followed by CB002 treatment of 16 hrs. B. DR5 was knocked-down by siRNA in DLD-1 cells followed by CB002 treatment of 16 hrs. C. NOXA was knocked-down by shRNA in SW480 cells followed by CB002 treatment of 16 hrs. D-E. Parental SW480 and SW480 NOXA stable knockdown cells treated with CB002 for 48 hrs were subjected to a sub-G1 analysis. E. Two-way ANOVA statistical analysis was performed for results from panel D, p ≤ 0.0001 against DMSO vehicle control. Three replicates were performed, and a representative histogram is shown. c-caspase 3 corresponds to the cleaved form of full-length caspase-3.
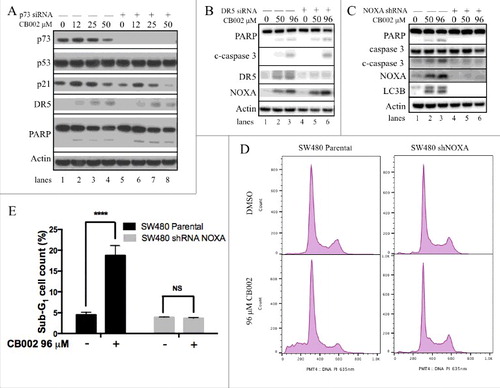
To confirm that cell death was mediated through apoptosis, we assessed the protein expression of PARP cleavage and cleaved caspases. Upon CB002 treatment, cleaved PARP and cleaved caspase-3 expression was observed to increase upon CB002 treatment as compared with vehicle control (, compare lane 1 to 2, 3, and 4; , compare lane 1 to 2 and 3). Taken together, these data indicate that CB002 induces the expression of p53 target genes and cell death in the SW480 cell line. Similar results were observed in DLD-1 cells (data not shown). To investigate the mechanism of CB002 in restoring the p53 pathway, we transiently knocked down p73 protein expression using siRNA (, see lanes 5–8) and probe for p53 target gene expression. We found an increase in the expression of p53 target genes p21 and DR5, and apoptosis associated marker PARP cleavage in p73-knockdown cells (, compare lane 5 to 6, 7, and 8). Overall these data suggest that p73 may not play a critical role in the mechanism of CB002-mediated p53 pathway restoration or cell death. In addition, CB002 treatment groups showed constant p53 protein expression levels as the DMSO control (, compare lane 1 to 2, 3, and 4). This indicates that CB002 might not have an effect on mutant p53 protein expression in SW480 cells. Nonetheless, further experiments were performed to corroborate this finding suggest some degradation effects toward certain p53 mutants (see description of ).
Figure 6. CB002 does not reduce mutant p53 stability in SW480 and DLD-1 cells. A-B. Experiments with SW480 cells. C. Analysis of DLD-1 colorectal cancer cells. Cells were treated for a 24 hr period with DMSO, CB002 or positive control followed by 100 μg/mL cycloheximide addition, and protein stability was evaluated in a time course ranging from 0 - 10 hrs. Histone deacetylase inhibitor, SAHA and Hsp90 inhibitor Geldanamycin (1 μM GA) were used as positive controls.

NOXA protein is required for CB002-induced cell death of tumor cells
To determine the role of p53 target genes DR5 and NOXA in CB002-mediated cell death, DR5 was knocked down by siRNA and a SW480 cell line stably transfected with NOXA shRNA plasmid construct was generated. Adequate DR5 knockdown was achieved as shown in (refer to lanes 4–6). DR5 knockdown in SW480 cells treated with 50 and 96 μM CB002 continued to induce the expression of cleaved-caspase 3 (c-caspase 3) and cleaved PARP ( compare lane 4 to 5 and 6) as efficiently as the scrambled siRNA-treated cells ( compare lanes 2 and 3 to 5 and 6). Therefore, DR5 appears dispensable for CB002-mediated cell death in the tumor cell lines that were tested.
By contrast, when NOXA was efficiently knocked down ( lanes 4–6), 50 and 96 μM CB002 treatment did not induce the expression of apoptotic markers cleaved caspase-3 and cleaved PARP (see lanes 5–6). We verified the requirement for NOXA in tumor cell apoptosis induction after exposure to CB002 by conducting sub-G1 analyses. As expected, 96 μM CB002 treatment in parental SW480 cells caused an increase in sub- G1 content (19%) as compared with DMSO treatment (4.5%). However, 96 μM CB002 treatment in SW480 where NOXA was stably knocked-down, failed to increase the content of sub-G1 cells (3.7%) when compared with DMSO treatment (3.93%) (). These data denote NOXA as a key mediator in the mechanism of action of CB002-mediated cell death, in the tumor cell lines tested.
In addition to CB002 increasing the expression of apoptotic markers, it was found to induce the expression of LC3B, a marker of autophagy ( compare lane 1 to 2 and 3). NOXA knock-down cells treated with 50 and 96 μM CB002 failed to induce LC3B expression ( see lane 5 and 6), indicating that NOXA is required for autophagy induction. The role of autophagy is further addressed below ().
CB002 treatment destabilizes the R175H p53 Mutant in tumor cells
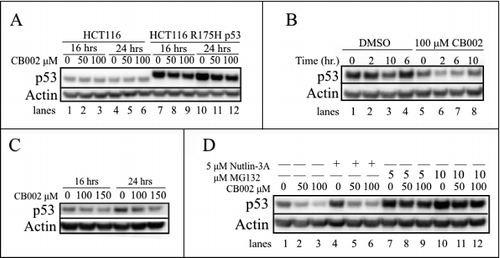
As mutant p53 can acquire a gain-of-function activity, targeting mutant p53 for degradation has been explored as a therapeutic strategy.Citation3,Citation10-16 We observed a constant mutant p53 protein expression in SW480 cells treated with CB002 for 16 hrs (, compare lane 1 to 2, 3, and 4). To confirm these findings, a cycloheximide chase assay was performed. Cells were treated with vehicle control or CB002 for a period of 24 hrs. Subsequently, 100 μg/mL cycloheximide was added to the wells and p53 stability was assessed at time points between 0 - 10 hrs. CB002 treatment did not affect mutant p53 stability in SW480 and DLD-1 cells (, compare lanes 1–5 to 6–10).
Nonetheless, CB002 treatment at 50 and 100 μM in HCT116 p53-null cells that exogenously expressed the R175H p53 mutant showed a decrease in mutant p53 protein expression compared with the DMSO control at 16 and 24 hrs ( compare lanes 7 to 8 and 9, and 10 to 11 and 12). To validate this result, we performed a cycloheximide chase experiment, as shown in . Using 100 μM CB002 treatment decreased the stability of mutant p53 compared with the vehicle control ( compare lanes 1–4 to 5–8). To investigate that the data observed was not an effect that is specific for exogenously expressed protein, RXF393 renal cancer cells that endogenously express R175H p53 were treated with 100 and 150 μM CB002. Both concentrations of CB002 at 16 and 24 hrs were found to decrease R175H p53 mutant expression as compared with the vehicle control (). Altogether, the data suggests that CB002 is capable of decreasing mutant p53 expression but potentially in a mutation-selective manner. In addition, CB002 p53 degradation was specific to mutant p53 as it was unable to decrease the expression of wild-type p53 (, lanes 1–6).
Figure 7. CB002 treatment reduces the stability of the R175H p53 mutant. A. CB002 reduces the protein expression of the exogenous R175H mutant in HCT116 p53-null cells and not the HCT116 wild-type p53 cells. B. HCT116 R175H p53 cells were treated for 24 hrs with DMSO or CB002 followed by 100 μg/mL cycloheximide addition, protein stability was evaluated from 0 - 10 hrs. C. CB002 reduces the protein expression of the endogenous R175H mutant p53 in RXF393 renal cancer cells. D. Co-treatment of 24 hrs with proteasome inhibitor MG132 and CB002 rescues the expression of the R175H mutant p53, suggesting the involvement of the ubiquitin proteasome system in CB002-mediated mutant p53 degradation.
CB002-induced R175H mutant p53 degradation in tumor cells is rescued by MG132
Mutant p53 can be degraded by various mechanisms including via MDM2-mediated degradation through the ubiquitin proteasome system. To explore the route by which the R175H p53 mutant was being degraded in response to CB002, HCT116 p53 R175H cells were pretreated with the MDM2 inhibitor Nutlin-3A and the proteasome inhibitor MG132 before CB002 treatment. After 1 hr of incubation, cells were simultaneously treated with CB002 and Nutlin-3A or MG132. Co-treatment using 50 or 100 μM CB002 along with Nutlin-3A for 24 hrs still resulted in reduction of R175H p53 mutant protein expression (, compare lane 4 to 5 and 6). These results suggest that MDM2 may not be required for the decreased R175H mutant p53 stability in the presence of CB002. On the other hand, treatment with CB002 and 2 different concentrations of MG132 (5 and 10 μM) appeared to rescue R175H mutant p53 protein expression (, compare lanes 7 to 8 and 9, and 10 to 11 and 12). Our data suggest that the R175H mutant p53 protein is degraded in human cancer cells in response to CB002 in a manner that is dependent on the ubiquitin proteasome system.
Autophagy does not play a role in mutant p53 degradation but it appears to be required for apoptotic cell death
During CB002 treatment, we noted that cells formed vacuoles suggesting an involvement of an autophagy process (). In addition, because it has been recently reported that mutant p53 can be degraded through the lysosome, we investigated whether CB002 may induce autophagy as a potential mechanism for R175H mutant p53 degradation. Using an autophagy detection kit (CYTO-ID® autophagy detection kit, Enzo® Life sciences) we observed an increase in autophagic vacuole-specific staining upon 50 μM CB002 treatment compared with the DMSO control in DLD-1 cells (). To confirm these findings, we probed a western blot for accumulation of LC3B, a well-known marker used in autophagy studies.Citation17 Because accumulation of LC3B can indicate a blockage of autophagy, LC3 conversion was evaluated during a time course from 0 - 48 hrs of treatment with 160 μM CB002 in DLD-1 cells. shows that following CB002 treatment there is an initial increase of LC3B and not in the DMSO treatment (compare lane 1 (0 hr) to lane 2, and 8). LC3B expression increases to a maximum in 8 hours of CB002 treatment (, lane 10) followed by a drop of its protein expression. No significant change in expression of LC3B was observed over time in DMSO control treated cells (, lanes 2–7). LC3B initially increases in the process of autophagy, and later it is degraded in the lysosomes, which is consistent with our observations. Thus, our data indicates that autophagy is induced by CB002 treatment.
Figure 8. CB002-mediated autophagy contributes to apoptotic cell death in drug-treated cells. CB002 induces autophagy as indicated by A. Specific recognition of autophagic vacuoles (detection using the CYTO-ID® autophagy detection kit at 24 hrs) and B. LC3B protein expression levels in DLD-1 cells (cells were treated with 160 μM CB002 or DMSO control for a period of 2–48 hrs. C. Autophagy inhibition by Chl does not rescue mutant p53 protein expression in HCT116 R175H p53 D. Blocking the autophagy process in SW480 cells with 50 μM chloroquine (Chl) reduces the expression of apoptotic markers cleaved caspase 8 (c-Caspase 8) and cleaved PARP (24 hrs) E. Inhibition of autophagy by Atg5 siRNA completely abolishes PARP cleavage in SW480 cells. F. Atg5-FADD apoptosis axis is not involved in CB002 mediated apoptosis in SW480 cells (24 hrs).
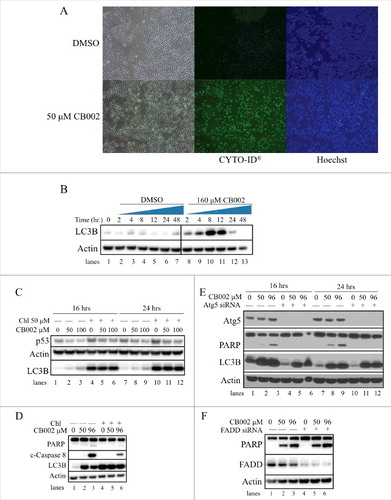
To explore if the R175H mutant p53 was being degraded through the autophagy mechanism, autophagy was blocked using the autophagosome/lysosome fusion inhibitor, chloroquine (Chl). As expected, treatment with CB002 in HCT116 p53 R175H cells for 16 and 24 hrs decreased mutant p53 expression (, compare lanes 1 to 2 and 3, and 7 to 8 and 9). Co-treatment with CB002 and Chl for 16 and 24 hrs did not rescue R175H mutant p53 protein expression, indicating that autophagy does not play a role in CB002 mediated R175H mutant p53 degradation (, compare lanes 4 to 5 and 6, and 10 to 11 and 12).
As autophagy can be induced for cell survival during cellular stress, we investigated its contribution to CB002-induced cell death. Autophagy was blocked using Chl added to CB002 to treat cells and the effects on apoptotic cell death markers were evaluated. CB002 induced the expression of cleaved caspases and PARP (, lanes 2 and 3). Upon combination treatment using CB002 and Chl, cleaved caspase 8 and cleaved PARP were reduced compared with CB002 alone (, compare lanes 2 and 3 to 5 and 6). To further validate this result, we downregulated autophagy by efficient siRNA-mediated knockdown of the autophagy related 5 (atg5) gene (, lanes 4–6, and 10–12). Ablation of atg5 resulted in complete loss of PARP cleavage upon treatment of CB002 for 16 and 24 hrs (, lanes 5–6 and 11–12). Although autophagy is mostly thought to be an adaptive process allowing the cell to survive during stress, here it seems to be required for CB002 mediated apoptotic cell death. Other investigators have demonstrated atg5 is implicated in autophagic cell death induced by IFN-γ via interaction with Fas-associated protein with death domain (FADD), a particular scenario that requires caspases.Citation18 Since our data suggests that atg5 is required for apoptosis, we then explored if the atg5-FADD axis was an interaction required for CB002 mediated cell death. As shown in lane 4–6, suitable knockdown of FADD was achieved. CB002 treatment was able to induce the expression of cleaved PARP in FADD knockdown cells ( lane 5 and 6) as efficient as the scrambled siRNA ( lane 2 and 3). Thus, FADD is not crucial in CB002 mediated cell death.
Discussion
In line with the fact that p53 is highly mutated in human tumors, strategies involving the restoration of wild-type conformation, mutant p53 degradation, and/or activation of p53 family members have been pursued and are of great interest for cancer therapy. Our current study identified and characterized a novel small molecule that restores the p53 pathway in various cancer cell lines that harbor mutant p53 protein.
The ability of CB002 to restore the p53 pathway was validated by documenting the activation of p53 target genes p21, DR5, and NOXA in various cancer cell lines and expression of well-established apoptotic markers. Stable knockdown of NOXA completely abrogated the expression of cleaved PARP and reduced sub-G1 cells, strongly implicating the relevance of NOXA as the key mediator of cell death induction by CB002. Although CB002 has a wide therapeutic index, it is important to note that CB002 was specific in causing apoptosis in cancer but not in normal cells. Given the selective anti-cancer activity of CB002, it is of interest as a tool compound and a candidate for future testing in different in vivo CRC mouse models. Our follow-up work involves further medicinal chemistry modifications to CB002 that enhance its therapeutic potency. Although its activity against cancer cells is in the micromolar range, this concentration range is not very different from what is required in cell culture with other FDA approved drugs such as sorafenib.
CB002 did not reduce the stability of the R273H/P309S (SW480) and S241F (DLD-1) p53 mutants but decreased the stability of the R175H p53 mutant (either endogenous or exogenously expressed in tumor cells). Reactivating the mutant p53 structure to the wild-type conformation strategy has been previously explored with agents such as CP31398Citation24 or others. Some p53 pathway-restoring small molecules in clinical trials may act via this mechanism.Citation15 Similar to the mutant-selectivity of CB002 observed in our present study, some other small molecules affect certain types of p53 mutants but not others. For example, Yu et al. discovered that small molecule NSC319726 is capable of reactivating the wild-type p53 conformation only in cells carrying the R175H p53 mutant. Its mutant-selectivity is impaired by the zinc ion chelating properties of the molecule.Citation12 Likewise, others have reported molecules that bind to specific pockets within the mutant p53 protein i.e. PhiKan083, and WR1065.Citation19,Citation16 Although some compounds have been found to be effective in reactivating the conformation of the mutant to the wild-type structure in a variety of p53 mutants, it is becoming clear that not all mutants act through the same mechanism.Citation3
Mutant p53 can be degraded through various cellular degradation routes. Our data suggests the R175H mutant is being degraded through the ubiquitin proteasome system independently of MDM2. Other ubiquitin ligases have been reported to mono-ubitquitinate p53 independently of MDM2 for instance, p53-induced protein with RING-H2 domain E3 (Pirh2), ARF-BP1, C-terminus of HSP70-interacting protein (CHIP), and caspase 8/10 associated RING proteins (CARPs).Citation20 Therefore, these and other regulatory proteins remain to be explored for any potential involvement in CB002-mediated R175H p53 mutant degradation.
Macro-autophagy and chaperone-mediated autophagy may impact on p53 protein turnover. Because we observed increased autophagic activity, we examined if these processes have any effect on R175H p53 mutant protein expression. Co-treatment of CB002 with Chl, a lysosome inhibitor, did not rescue mutant p53 protein expression, indicating that these processes are not involved in CB002 mediated R175H p53 mutant protein degradation. Interestingly, confirming these results by inhibiting autophagy through atg5 knockdown, led us to the finding that atg5 was required for apoptosis, as indicated by cleaved PARP. Atg5, mostly known for its role during the initial steps of autophagosome development, has been shown to be involved in autophagic cell death. This form of cell death is caspase-independent and characterized by the appearance of double membrane cytoplasmic vesicles whose cargo is degraded by the lysosome. Others have demonstrated atg5 to be implicated in autophagic cell death induced by IFN-γ via interaction with FADD, a particular scenario that required caspases.Citation18 Since our data suggested that atg5 is required for apoptosis, we then explored if the atg5-FADD axis was an interaction required for CB002 mediated cell death. Knockdown of FADD did not affect PARP cleavage, indicating that FADD is not crucial in CB002-mediated cell death. Furthermore, atg5 has been recognized as a molecular substrate of calpain that shifts autophagy to apoptosis.Citation21 Here, the truncated atg5 form generated by calpain cleavage was demonstrated to translocate into the mitochondria ultimately leading to cytochrome c release and caspase activation. From these data we could speculate that the truncated form of atg5 is responsible for CB002-mediated cell death. To test this hypothesis in the future, calpain inhibitors would be useful to delineate the importance of truncated atg5 in apoptosis. It is important to note that atg5 siRNA mediated knockdown still showed an increase in LC3B expression levels, indicating that the autophagy process was not hindered (). Since the process of autophagy has been reported to occur independently of atg5,Citation22 more experiments need to be done to directly implicate the role of the autophagy process in cell death rather than the atg5 protein itself, even thought blockage with Chl had an effect on cell death.
Our study identified CB002 as a novel p53 pathway-restoring compound that induces apoptosis in cancer cell lines, mainly through NOXA expression. NOXA shRNA-mediated knockdown prevented cells from undergoing apoptosis, and interestingly, largely eliminated LC3B expression (). NOXA has been thought to play an anti-apoptotic role by inducing autophagy, and thus, delaying apoptosis.Citation23 Nonetheless, our results suggest that autophagy contributes to apoptosis and that autophagy induction might be dependent on NOXA expression (). Even though our findings suggest that only the stability of the R175H p53 mutant was affected, we cannot assert complete selectivity on this mutant, as a larger panel of p53 mutants should be further tested. Furthermore, degradation of mutant p53 does not seem to be required for p53 pathway restoration, as CB002 was able to induce several p53 target genes in various mutant p53 cell lines without having any impact in their p53 protein turnover. In summary, CB002 is a novel p53 pathway-restoring compound that induces tumor selective cell death through NOXA induction, and can target certain mutants of p53 such as the R175H mutant for MDM2-independent proteasomal degradation. Further studies are required, including in vivo studies, to determine the anti-cancer effects of CB002 or analogs. CB002 may be further developed as a cancer therapeutic.
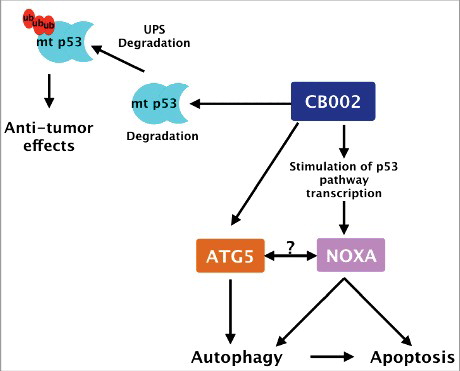
Figure 9. CB002 proposed mechanism of action. CB002 induces the expression of NOXA and the degradation of mutant p53 through the ubiquitin proteasome system. NOXA and atg5 expression are required for apoptotic cell death. The relationship between ATG5 and NOXA, if any, remains to be determined. Furthermore, CB002 induces autophagy which aids apoptotic cell death in drug-treated cells.
Materials and methods
Cell-based drug screening for p53 pathway-restoring small molecules
High-throughput screening was performed using a non-invasive bioluminescence imaging in human colorectal cancer cell lines that stably express a p53-regulated luciferase reporter.Citation7 Cells were seeded in 96-well plates (Greiner Bio-One) at a density of 1 × 104 cells per well. p53 transcriptional activity was imaged using an IVIS imaging system for a period of 3–24 hrs.
Cell lines and culture conditions
DLD-1, SW480, HCT116, and HCT116 p53−/− colorectal cancer cell lines that stably express a p53-regulated luciferase reporter were previously generated in our laboratory.Citation7 RXF393 renal cancer cell lines, and WI38 and MRC5 normal lung cell fibroblasts were purchased from ATCC. Cell lines were maintained in HyClone™ Dulbecco's High Glucose Modified Eagles Medium (DMEM, GE Healthcare), HyClone™ McCoy's 5A (GE Healthcare), HyClone™ RPMI 1640 (GE Healthcare), or Eagle's Minimum Essential Medium (EMEM, ATCC) containing 10% fetal bovine serum and 1% penicillin/streptomycin (complete media) at 37°C in 5% CO2, as recommended by ATCC.
CellTiter-Glo® luminescent cell viability assay
Cells were seeded in 96-well plates at a density of 5 × 103 cells per well. 20 μL of CellTiter-Glo® Reagent was added directly to the wells, following the manufacturer's protocol, and bioluminescence signal was determined using an IVIS imaging system.
Knockdown expression of p73, DR5, ATG5, and FADD using siRNA
A total of 1 × 105 cells/well were plated per well in a 12-well plate in medium with 10% FBS without antibiotic. Forward transfection of p73 siRNA (s14319, Ambion®), DR5 (sc-40237, Santa Cruz Biotechnology), atg5 (137766, Ambion®), FADD (S16706, Ambion®) was performed using the Lipofectamine® RNAiMAX Transfection Reagent (Life Technologies) and incubated for 48 hrs before treatment.
Overexpression of p53 R175H mutant by lentivirus infection
HCT116 p53−/− cells (previously obtained from the Vogelstein Laboratory, Johns Hopkins University) were infected with a lentivirus vector containing the p53 R175H mutant (pLenti6/V5-p53_R175H, Addgene). Cells were selected with blasticidin (8 μg/mL) containing media cultured for 10 d. Blasticidin-resistant clones (pooled clones) were screened for expression of the p53 R175H mutation by western blot analysis with p53 DO-1 antibody.
Knockdown expression of NOXA by lentivirus infection
A NOXA shRNA plasmid construct was amplified according to the manufacturer's recommendation (TRC Lentiviral Human PMAIP1 shRNA, Dharmacon). Plasmid DNA was isolated using the PureLink® HiPure Plasmid Filter Maxiprep Kit (Invitrogen) according to the manufacturer's instructions. Lentivirus production was performed by transfecting HEK293T cells at a density of 8 × 106 cells per 10 cm dish with 1.6 μg pMD2.G envelope plasmid, 3.2 μg psPAX2 packaging vector, 3.2 μg plasmid DNA, and 24 μL of lipofectamine® Transfection reagent 2000 (Life Technologies) in a total volume of reaction of 1 mL of antibiotic free DMEM media for a period of 6–10 hrs. Media was then replaced with antibiotic free DMEM. Lentiviral particles were collected between 48–72 hrs. SW480 cells (2.3 × 106 cells per well in a 12-well plate) were infected 1:1 (virus containing media: antibiotic free DMEM media, total volume 1 mL) for a period of 24 hrs. Then, media was replaced with DMEM complete media for an additional 24 hrs. At this point, cells were split and seeded (20% confluent) for selection in a 10 cm dish with puromycin (2.5 μg/mL)-containing complete DMEM media and cultured for 10 d. Puromycin containing complete media was replaced every 2–3 d. Puromycin-resistant clones were screened for knockdown of NOXA by Western Blot analysis with NOXA antibody.
Colony formation assay
Cells were seeded in 6-well plates at a density of 500 cells per well. Cells were treated with CB002 small molecule for 24 hrs. Then, cells were cultured in drug-free complete media for 15 d. During the course of 15 days, the media was changed every 2–3 d. At the end of the 2 weeks, media was removed, wells were washed twice with Dulbecco's phosphate buffered saline (PBS) and the colonies were fixed and stained with 10% methanol and 0.25% crystal violet (Sigma-Aldrich) for 30 min. Wells were then carefully washed with distilled and deionized water and allowed to dry.
Apoptosis assay
Apoptotic cells were quantified by sub-G1 analysis. Cells were seeded at a density of 2.5 × 105 – 5 × 105 in a 6-well plate and treated for 48–72 hrs. After treatment, adherent cells were trypsinized and collected along with floating cells, washed with PBS and fixed in 70% ethanol. Cells were then incubated in a Phosphate-citric acid buffer (0.2 M Na2HPO4 + 0.1 M Citric Acid, pH 7.8) at room temperature for 5 min, spun down and resuspended for staining with 50 μg/mL propidium iodide (PI) in the presence of 250 μg/mL pancreatic ribonuclease (RNase A). Sub-G1 analyses were performed using an Epics Elite Epics flow cytometer (Beckman-Coulter).
Immunoblotting
After treatment, cells were harvested by trypsinization, washed with PBS, and lysed with RIPA buffer (Sigma-Aldrich) for 30 min – 1 hr at 4 ºC. Protein lysates were spun down and supernatant was collected. Protein quantification was performed using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). 1 x NuPAGE® LDS sample buffer (Thermo Fisher Scientific) and 2-Mercaptoethanol as the reducing agent (Sigma-Aldrich) were added to protein lysates, followed by boiling for 15 min at 95 ºC. Equal total protein amounts samples were loaded into NuPAGE™ Novex™ 4–12% Bis-Tris Protein Gels (1.5 mm, Thermo Fisher Scientific) and gel eletrophoresis was performed with NuPAGE™ MES SDS Running Buffer. Proteins were transferred onto an Immobilon-P membrane (PVDF, EMD Millipore) using a Bio-Rad system with a 10% Tris-Glycine and 10% methanol transfer buffer diluted in distilled and deionized water. After transfer, membranes were blocked with 10% milk in TBST solution and then incubated overnight with primary antibody, washed with TBST and incubated with secondary antibody for 1 hr. Incubations were performed in 5% milk in TBST solution. Signal was detected by using a chemiluminescent detection kit, followed by autoradiography. The following antibodies were used: p53 (DO-1, 1:1000, Santa Cruz), p73 (1:1000, Bethyl Laboratories), p21 and NOXA (1:250, EMD Millipore), DR5, FADD, cleaved caspase 3, cleaved caspase 8, cleaved PARP, and LC3B (1:1000, Cell Signaling), and β-actin (1:10000, Sigma).
Statistical analysis
Data are presented as means ± SEM (3 biologic replicates). To assess the statistical significance of the differences, Two-way ANOVA for 2 comparisons was performed, with p < 0.05 defined as statistically significant. Comparisons were made against the DMSO vehicle control.
Disclosure of potential conflicts of interest
W.S.E-D. is a Founder of p53-Therapeutics, Inc., a biotech company focused on developing small molecule anti-cancer therapies targeting mutant p53. W.S.E-D. has disclosed his relationship with p53-Therapeutics and potential conflict of interest to his academic institution/employer and is fully compliant with NIH policies and institutional policies that is managing this potential conflict of interest.
Acknowledgments
This work was presented in part at the annual American Association for Cancer Research (AACR) meetings in 2015, 2016, and 2017. L.J.H-B. received the AACR Minority Scholar Research Award in 2017.
Additional information
Funding
References
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9(5):402–12; PMID:18431400; https://doi.org/10.1038/nrm2395
- Hong B, van den Heuvel AP, Prabhu VV, Zhang S, El-Deiry WS. Targeting tumor suppressor p53 for cancer therapy: Strategies, Challenges and Opportunities. Curr Drug Targets 2014; 15(1):80–9; PMID:24387333; https://doi.org/10.2174/1389450114666140106101412
- Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014; 25(3):304–17.
- Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ. Gain of function mutations in p53. Nat Genet 1993; 4(1):42–6; PMID:8099841; https://doi.org/10.1038/ng0593-42
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119(6):861–72; PMID:15607981; https://doi.org/10.1016/j.cell.2004.11.006
- Oshima Y, Sasaki Y, Negishi H, Idogawa M, Toyota M, Yamashita T, Wada T, Nagoya S, Kawaguchi S, Yamashita T, et al. Antitumor effect of adenovirus-mediated p53 family gene transfer on osteosarcoma cell lines. Cancer Biol Ther 2007; 6(7):1058–66; PMID:17568187; https://doi.org/10.4161/cbt.6.7.4320
- Wang W, Kim SH, El-Deiry WS. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci U S A 2006; 103(29):11003–8; PMID:16835297; https://doi.org/10.1073/pnas.0604507103
- Hong B, Prabhu VV, Zhang S, van den Heuvel AP, Dicker DT, Kopelovich L, El-Deiry WS. Prodigiosin rescues deficient p53 signaling and anti-tumor effects via up-regulating p73 and disrupting its interaction with mutant p53. Cancer Res 2014; 74(4):1153–65; PMID:24247721; https://doi.org/10.1158/0008-5472.CAN-13-0955
- Zhang S, Zhou L, Hong B, van den Heuvel AP, Prabhu VV, Warfel NA, Kline CL, Dicker DT, Kopelovich L, El-Deiry WS. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res 2015; 75(18):3842–52; PMID:26294215; https://doi.org/10.1158/0008-5472.CAN-13-1079
- Vakifahmetoglu-Norberg H. Chaperone-mediated autophagy degrades mutant p53. Genes Dev 2013; 27(15):1718–30; PMID:23913924; https://doi.org/10.1101/gad.220897.113
- Garufi A. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis 2014; 5:e1271; https://doi.org/10.1038/cddis.2014.217
- Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell 2010; 21(5):614–25
- Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 2002; 8(3):282–8; PMID:11875500; https://doi.org/10.1038/nm0302-282
- Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ 2011; 18(12):1904–13; PMID:21637290; https://doi.org/10.1038/cdd.2011.71
- Parrales A, Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol 2015; 5:288; PMID:26732534; https://doi.org/10.3389/fonc.2015.00288
- North S, Pluquet O, Maurici D, El-Ghissassi F, Hainaut P. Restoration of wild-type conformation and activity of a temperature-sensitive mutant of p53 (p53(V272M)) by the cytoprotective aminothiol WR1065 in the esophageal cancer cell line TE-1. Mol Carcinog 2002; 33(3):181–8; PMID:11870884; https://doi.org/10.1002/mc.10038
- Mizushima N, Yoshimori T. How to Interpret LC3 Immunoblotting. Autophagy 2007; 3(6):542–5; PMID:17611390; https://doi.org/10.4161/auto.4600
- Pyo JO. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005; 280(21):20722–9; PMID:15778222; https://doi.org/10.1074/jbc.M413934200
- Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci USA 2008; 105(30):10360–165; PMID:18650397; https://doi.org/10.1073/pnas.0805326105
- Vinod P, Guillermina L. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev 2014; 28(16):1739–51; PMID:25128494; https://doi.org/10.1101/gad.247452.114
- Yousefi S. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8(10):1124–32; PMID:16998475; https://doi.org/10.1038/ncb1482
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461 (7264):654–8; PMID:19794493; https://doi.org/10.1038/nature08455
- Liu YL. Noxa upregulation by oncogenic activation of MEK/ERK through CREB promotes autophagy in human melanoma cells. Oncotarget 2014; 5(22):1237–51.
- Takimoto R, Wang W, Dicker DT, Rastinejad F, Lyssikatos J, el-Deiry WS. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol Ther 2002; 1(1):47–55; https://doi.org/10.4161/cbt.1.1.41