
ABSTRACT
Mycobacterium tuberculosis (M.tb), which causes tuberculosis, is a host-adapted intracellular pathogen that can live within macrophages owning to its ability to arrest phagolysosome biogenesis. The guanine nucleotide exchange factor H1 (GEF-H1) may contribute to the phagocytosis of bacteria by macrophages through mediating the crosstalk between microtubules and the actin cytoskeleton. Its role in Shigella infection has been determined but little is known about the role of GEF-H1 in mycobacterial infection. In the present study, we demonstrated that GEF-H1 functioned as a key regulator of the macrophage-mediated anti-mycobacterial response. We found that both mRNA and protein expression levels of GEF-H1 were significantly upregulated in macrophage during mycobacterial infection. Moreover, silencing of GEF-H1 with specific siRNAs reduced the phosphorylation of p38 mitogen-activated protein kinase and TANK binding kinase 1 as well as the expression of interleukin-1β (IL-1β), IL-6, and interferon-β (IFN-β), without affecting nitric oxide production or autophagy. Importantly, GEF-H1 depletion attenuated macrophages-mediated mycobacterial phagocytosis and elimination. Taken together, our data supported that GEF-H1 was a novel regulator of inflammatory cytokine production and mycobacterial elimination, and may serve as a novel potential target for clinical treatment of tuberculosis.
Introduction
Mycobacterium tuberculosis (M.tb) is the causative agent of tuberculosis (TB). In 2016, about 10.4 million people acquired TB, and 1.5 million people died from this disease.Citation1 Approximately 30% of exposed individuals will become tuberculin-positive patients, although only 5–10% of these infected individuals will develop clinical manifestations of active TB. In contrast, the majority of infected individuals developed latent TB, in which the invading M.tb can survive asymptomatically within host macrophages for a long period.Citation2
Macrophages, which internalize both pathogenic and nonpathogenic mycobacteria, are the primary phagocytic cells involved in the control of M.tb infection. While serving as the host cell niche for M.tb, macrophages also promote the clearance of M.tb through finely tuned immune responses. Upon recognition of the invading M.tb by pattern recognition receptors (PRRs), macrophages first activate intracellular signaling cascades, including the pathways of mitogen-activated protein (MAP) kinase (MAPK) and TANK binding kinase-1 (TBK1), and subsequently induce the production of cytokines, such as interleukin (IL)-1β, IL-6 and IFN-β.Citation3 In addition, macrophages use myriad defense mechanisms to fight against the invading M.tb, including nitric oxide (NO) production, protease activation, and autophagy induction.Citation4,Citation5
Guanine nucleotide exchange factor H1 (GEF-H1), also called lfc in mice, is encoded by the arhgef2 gene.Citation6 GEF-H1 was originally identified as a member of the Dbl family directs spatio-temporal activation of Rho GTPases.Citation7 Inactive GEF-H1 binds to the dynein motor complex on microtubules (MTs),Citation9,Citation10 in its MT-bound state, the guanosine-exchange activity of GEF-H1 is suppressed; in contrast, when GEF-H1 is released from MTs, its Rho-specific GEF activity is activated.Citation8 Rho GTPases act as molecular switches to control many basic cellular activities and are critical for several specialized cellular functions, such as actin cytoskeleton organization, cell migration and adhesion, reactive oxygen species formation, and apoptosis.Citation8 GEF-H1 is also known to mediate the crosstalk between MTs and the actin cytoskeleton,Citation11 which may contribute to the phagocytosis of bacteria by macrophages. Moreover, lipopolysaccharide-induced nuclear factor (NF)-κB translocation and activation are inhibited by GEF-deficiency,Citation8 and GEF-H1 also contributes to pathophysiological NF-κB signaling involved in Shigella infection in the intestinal epithelium.Citation12 However, the role of GEF-H1 in macrophage-mediated immune responses against mycobacteria remains unclear.
Accordingly, in the present study, we evaluated the role of GEF-H1 in the macrophage-mediated antimycobacteria immune response. Our results provide important insights into the function of GEF-H1 in regulating the inflammatory cytokine response and mycobacterial elimination in macrophages during mycobacterial infection.
Results
Upregulation of GEF-H1 during mycobacterial infection
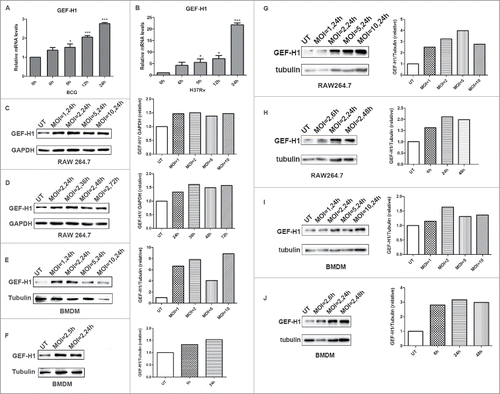
To determine the expression of GEF-H1 during mycobacterial infection, RAW264.7 cells were infected with BCG or H37Rv for the indicated time, and the expression level of GEF-H1 was detected. Real-time PCR results showed that GEF-H1 mRNA levels were significantly increased upon BCG or H37Rv infection ( and ). Similarly, western blot analysis revealed that the protein expression level of GEF-H1 was increased in RAW264.7 cells in response to BCG or H37Rv infection for the indicated MOI and time (, , , and ). Similarly, the expression of GEF-H1 in bone marrow-derived macrophages (BMDMs) was also upregulated upon BCG or H37Rv infection (, , , and ). Additionally, quantitative measurement results showed that GEF-H1 protein expression was altered independently of concentration and time; GEF-H1 expression reached a maximum at a multiplicity of infection (MOI) of 5 after 24 h. Thus, these results indicated that GEF-H1 expression was induced in response to mycobacterial infection in macrophages.
Figure 1. GEF-H1 expression increased in macrophage during mycobacterial infection. (A and B) RAW264.7 cells were infected with BCG (A) or H37Rv (B) for 0, 6, 9, 12, or 24 h, and GEF-H1 mRNA levels were then evaluated by real-time PCR. (C and D) RAW264.7 cells were infected with BCG at the indicated MOI (C) or for the indicated time (D), and GEF-H1 protein levels were evaluated by western blotting. (E and F) BMDMs were infected with BCG at the indicated MOI (E) or for the indicated time (F), and GEF-H1 protein levels were evaluated by western blotting. (G and H) RAW264.7 cells were infected with H37Rv at the indicated MOI (G) or for the indicated time (H), and GEF-H1 protein levels were evaluated by western blotting. (I and J) BMDMs were infected with H37Rv at the indicated MOI (I) or for the indicated time (J), and GEF-H1 protein levels were evaluated by western blotting (*, p < 0.05; ***, p < 0.001).
Mycobacterial infection induced GEF-H1 expression was dependent on the MAPK signaling pathway and siRNA-mediated knockdown of GEF-H1 in macrophages.
To investigate the mechanisms mediating the upregulation of GEF-H1 expression during mycobacterial infection, we pretreated RAW264.7 cells with the inhibitors SP600125 (c-Jun N-terminal kinase [JNK]), JSH23 (NF-κB), SB203580 (p38), and U0126 (extracellular signal-regulated kinase [ERK]) and then infected the cells with BCG for 24 h. Real-time PCR analysis showed that GEF-H1 mRNA was significantly downregulated following treatment with U 0126, with or without BCG infection (). Additionally, GEF-H1 mRNA and the protein expression level of GEF-H1 was significantly downregulated following pretreatment with SB 203580, JSH 23, or SP 600125 in BCG-infected cells ( and ).
Figure 2. Mycobacterial infection induced GEF-H1 expression was dependent on MAPK signaling pathway, and specific siRNA decreased GEF-H1 expression in macrophages. (A and B) RAW264.7 cells were pretreated with 10 μM SP600125, 300 μM JSH23, 50 μM SB203580, or 50 μM U0126 for 1 h and then infected with BCG for 24 h. (A) GEF-H1 mRNA was evaluated by real-time PCR. (B) GEF-H1 protein expression was evaluated by western blotting. (C and D) RAW264.7 cells (C) and BMDMs (D) were transfected with 100 nM Si-NC or siRNA for GEF-H1 for 30 h. GEF-H1 mRNA levels were evaluated by real-time PCR, and GEF-H1 protein levels were evaluated by western blotting (ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001).
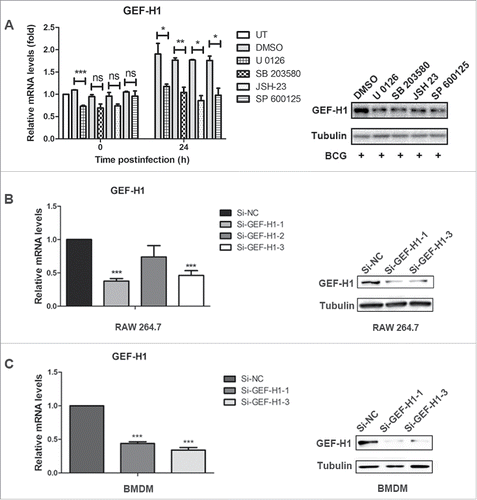
To definitively determine the role of GEF-H1 in regulating the inflammatory response in macrophages, we used GEF-H1-specific small interfering RNA (siRNA) to knock-down GEF-H1 in both RAW 264.7 cells and BMDMs. RAW264.7 cells and BMDMs were transfected with negative control (NC) siRNA (Si-NC) or GEF-H1-specific siRNA under optimized conditions, and the expression level of GEF-H1 was evaulated. Real-time PCR and western blot analysis showed that the mRNA and protein levels of GEF-H1 were significantly decreased after transfection with Si-GEF-H1–1 or Si-GEF-H1–3 ( and ).
GEF-H1 regulated macrophage cytokine expression in response to mycobacterial infection.
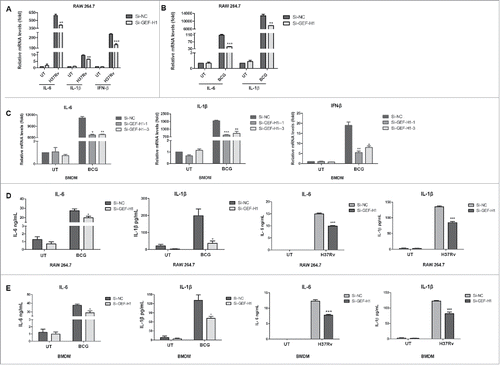
To determine whether GEF-H1 modulated cytokine production in macrophages in response to mycobacterial infection, RAW264.7 cells and BMDMs were transiently transfected with Si-NC or siRNA for GEF-H1, and then infected with H37Rv or BCG. Our analysis showed that GEF-H1 depletion in RAW264.7 cells significantly decreased IL-6, IL-1β, and IFN-β mRNA expressoin in response to H37Rv infection () or BCG infection (); similar results were obtained in BMDMs infected with BCG (). Next, RAW264.7 cells and BMDMs were transfected with Si-NC or Si-GEF-H1, and subsequently infected with H37Rv or BCG. Analysis of cytokine levels in cell-free culture supernatants by enzyme-linked immunosorbent assay (ELISA) showed that production of IL-6 and IL-1β was significantly decreased in both in RAW264.7 cells () and BMDMs () after GEF-H1 knockdown in response to either BCG or H37Rv infection.
Figure 3. GEF-H1 regulated M.tb- and BCG-mediated cytokines production and secretion. (A and B) RAW264.7 cells were transfected with 100 nM siRNA for 24 h and then infected with H37Rv (A) or BCG (B) at an MOI of 2 for 6 h. IL-6, IL-1β, and IFN-β expression levels were assessed by real-time PCR. (C) BMDMs were transfected with 100 nM siRNA for 24 h and then infected with BCG at an MOI of 2 for 6 h. IL-6, IL-1β, and IFN-β mRNA levels were then assessed by real-time PCR. (D and E) RAW264.7 cells (D) and BMDMs (E) were grown in 12-well tissue culture plates, transfected with siRNA for 12 h, and then incubated with BCG or H37Rv (MOI = 2) for 24 h. Supernatants were collected, and IL-6 and IL-1β levels were measured by ELISA (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
NO production and autophagy in response to mycobacterial infection upon GEF-H1 depletion.
In macrophages, NO production, which requires inducible nitric oxide synthase (iNOS) expression, is an important innate immune response against mycobacteria.Citation13,Citation14 To determine whether GEF-H1 mediated NO production in macrophages, RAW264.7 cells were transiently transfected with Si-NC or siRNA for GEF-H1 and subsequently infected with BCG. The results showed that BCG induced iNOS mRNA expression, but GEF-H1 depletion did not alter either iNOS mRNA expression or NO production ( and ).
Figure 4. GEF-H1 did not affect NO production or autophagy in macrophages. (A and B) RAW264.7 cells were transfected with Si-NC or siRNA for GEF-H1 for 30 h, and then infected with BCG (MOI = 2) for the indicated times. Real-time PCR was used to determine iNOS mRNA levels (A). Supernatants were collected, and NO production was tested by Griess assay (B). (C) RAW264.7 cells were transfected with Si-NC or siRNA for GEF-H1 for 30 h and then infected with BCG for 12 h. The amount of LC3 was determined by western blotting.
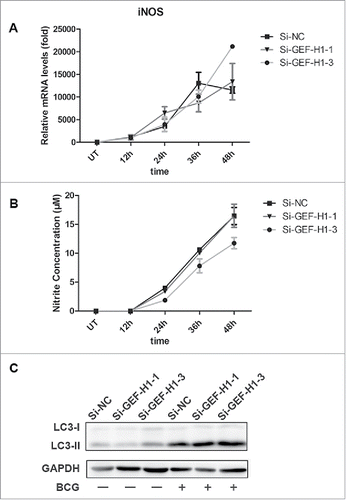
Autophagy has been shown to play an important role in the host immune response against mycobacterial infection in macrophage. Microtubule-associated protein light chain 3 (LC3) is a characteristic marker of autophagy.Citation15 To determine the role of GEF-H1 in autophagy of macrophages during mycobacterial infection, RAW264.7 cells were transiently transfected with Si-NC or siRNA for GEF-H1 and subsequently infected with BCG, the amount of LC3 was evaulated by western blotting. The results showed that silencing of GEF-H1 did not affect the amount of LC3 (), suggesting that GEF-H1 was not involved in modulation of autophagy in macrophages.
GEF-H1 promoted the activation of p38 MAPK and TBK1 in macrophage upon mycobacterial infection.
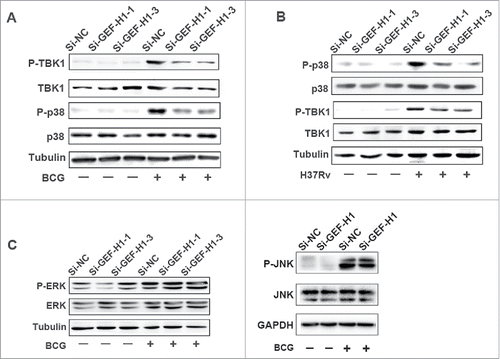
To further investigate the factors involved in GEF-H1-regulated cytokine production, we analyzed the phosphorylation of p38 MAPK, TBK1, ERK1/2, and JNK by western blotting after transient transfection of cells with Si-NC or GEF-H1 siRNA and infection with BCG. The phosphorylation levels of p38 MAPK and TBK1 were significantly decreased in cells transfected with Si-GEF-H1 in response to either BCG () or H37Rv infection (). However, the phosphorylation levels of ERK and JNK in GEF-H1-depleted cells were unaffected as compared with those in cells transfected with Si-NC (). These results suggested that GEF-H1 may promote cytokines expression by activating p38 MAPK and TBK1 signaling pathways but not ERK or JNK signaling pathways in macrophages during mycobacterial infection.
Figure 5. Silencing GEF-H1 decreased the phosphorylation of p38 and TBK1. (A and B) RAW264.7 cells were transfected with Si-NC or GEF-H1 siRNA and then infected with BCG (A) or H37Rv (B) for 0.5 h. Phosphorylation levels of p38 and TBK1 were analyzed by western blotting. (C) RAW264.7 cells were transfected with Si-NC or GEF-H1 siRNA and then infected with BCG for 0.5 h. Phosphorylation levels of ERK and JNK were analyzed by western blotting.
GEF-H1 depletion impaired macrophage-mediated phagocytosis and elimination of intracellular mycobacteria.
To investigate the role of GEF-H1 in macrophage phagocytic capacity during mycobacterial infection, we next examined its effects on mycobacterial viability by colony-forming unit (CFU) assay. RAW264.7 cells transiently transfected with Si-NC or Si-GEF-H1 were infected with BCG. Our analysis showed that GEF-H1 depletion significantly reduced the amount of intracellular BCG in RAW264.7 cells at indicated time points (). Additionally, the survival of intracellular BCG significantly increased in RAW264.7 cells at the indicated time points (). Which means GEF-H1 depletion impaired macrophage elimination of intracellular BCG.
Figure 6. Silencing of GEF-H1 decreased phagocytosis and intracellular BCG killing in RAW264.7 cells. RAW264.7 cells were transfected with Si-NC or Si-GEF-H1 for 30 h and then infected with BCG (MOI = 10) for 1 h. The intracellular viable bacilli were determined by CFU assays at the indicated time post-infection (A). Survival rate was calculated compared with that at 0 h (B). (C) RAW264.7 cells were transfected with Si-NC or Si-GEF-H1 for 30 h and then infected with Texas Red-labeled BCG at an MOI of 10 for 1 h. Phagocytosis of mycobacteria was determined by flow cytometry. (D) The mean fluorescence intensity (MFI) of the Texas red positive population was calculated using of FlowJo 7.6.1 software (*, p < 0.05; **, p < 0.01).
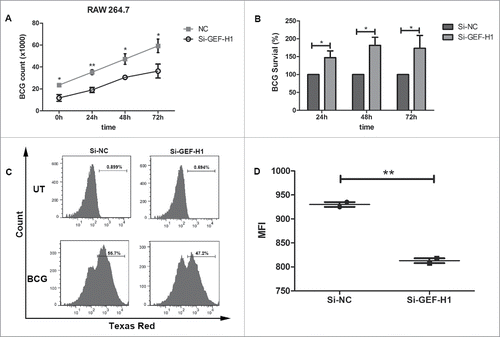
Furthermore, to further comfirm whether GEF-H1 affected mycobacterial phagocytosis by macrophages, RAW264.7 cells were transfected with Si-NC or Si-GEF-H1, infected with fluorescein Texas red-labeled BCG, and subjected to flow cytometry analysis. The data showed that GEF-H1 depletion reduced phagocytosis in RAW264.7 cells (). Moreover, the mean fluorescence intensity (MFI) of the Texas red positive population was significantly decreased in the group transfected with Si-GEF-H1 () compared with that in cells transfected with Si-NC. These results implied that GEF-H1 was invovled in macrophage-mediated phagocytosis and elimination of intracellular mycobacteria.
Discussion
M.tb is a pathogen that is difficult to eliminate and can take multiple steps to evade the host cells, such as macrophages.Citation16 M. bovis BCG is an attenuated strain of M. bovis lacking the RD-1 region, which contains important virulence determinants.Citation17,Citation18 However, it still shares many features with M.tb, such as lipoprotein expression.Citation19 As one of the most important cell types in antimycobacterial immunity, macrophages function as the predominant responder against mycobacterial infection. Thus, enhancement of the antibacterial properties of macrophages has been a major focus of many researchers. GEF-H1 serves as a gatekeeper on MTs to locally modulate the activity of GTPases, which in turn are responsible for the initial polarization of the MT cytoskeleton to facilitate antiviral responses.Citation20 However, the role of GEF-H1 in macrophages during mycobacterial infection has not been elucidated. In the present study, we investigated, for the first time, the role of GEF-H1 during mycobacterial infection. Our results showed that the expression of GEF-H1 was increased via the MAPK signaling pathway during mycobacterial infection. Silencing of GEF-H1 decreased the phosphorylation level of p38 MAPK and TBK1 and subsequently attenuated the expression of the inflammatory cytokines IL-1β and IL-6. More importantly, silencing of GEF-H1 inhibited macrophage-mediated mycobacterial phagocytosis and elimination. Collectively, our results indicated that GEF-H1 was a novel molecule regulating inflammatory cytokine response and mycobacterial elimination and may serve as a potential target for the clinical treatment of TB.
GEF-H1 appears to be a crucial adaptor in coupling MT dynamics to Rho GTPase activation in a variety of biologic processes. Additionally, numerous pathophysiological conditions result in changes in the integrity of the MT cytoskeleton, including epithelial cell and endothelial cell dysfunction and infectious disease.Citation21 Several studies have shown that GEF-H1 regulates the role of Rip2 in adaptive immune function and is required for successful immunoglobulin isotype switching after immunization with ovalbumin and for the production of IFN-γ in T-helper type 1 (Th1) cells and natural killer (NK) cells.Citation22,Citation23 In response to Shigella infection, GEF-H1 is enriched in bacterial entry sites in an actin-dependent manner, thereby promoting signal transduction; these results confirmed that GEF-H1 is upregulated in response to bacterial infection.Citation24 Moreover, Cullis J et al. found that GEF-H1 is a transcriptional target of the RAS/MAPK pathway.Citation25 In the present study, we found that GEF-H1 expression was increased in RAW 264.7 cells in response to H37Rv or BCG challenge. Furthermore, the expression of GEF-H1 was also increased in BMDMs during H37Rv or BCG infection. Moreover, after mycobacterial infection, MAPK pathway activation is essential for macrophage antimycobacteria immune response. We showed that GEF-H1 expression was dependent on the MAPK signaling pathway including ERK, p38 MAPK, NF-κB and JNK after mycobacterial infection. These findings suggested a potential correlation between GEF-H1 and mycobacterial infection.
The activation of macrophages leads to the production of various cytokines, including IL-1β and IL-6, which are important for orchestrating appropriate inflammatory and adaptive immune responses.Citation26 IL-1β is a potent cytokine required for host resistance to TB,Citation27,Citation28 and IL-6 is required for the optimal T-cell response against M. tb infection.Citation29 In our study, we demonstrated, for the first time, that production of IL-6 and IL-1β was decreased in both RAW 264.7 and BMDMs when GEF-H1 expression was silenced. Our results indicated that GEF-H1 was necessary to induce cytokines expression in macrophages during mycobacterial infection. Cytokine expression is regulated by various modulators, such as MAPKs, TBK1, and NF-κB.Citation30-33 Moreover, our results revealed that silencing GEF-H1 decreased the phosphorylation levels of p38 MAPK and TBK1, thus leading to reduced IL-6 and IL-1β release in macrophages after mycobacterial infection.
NO is an effective host defense molecule that protects against microbial pathogens. NO can damage DNA and interact with accessory protein targets to cause enzymatic inactivation.Citation34 Thus, we are interested in determining whether GEF-H1 regulates the release of NO form RAW 264.7 cells in response to BCG infection. Our data show that GEF-H1 failed to regulate the expression of iNOS mRNA and subsequent NO production in macrophages. As an important functions of macrophages, autophagy is an evolutionarily conserved process that is involved in maintaining cytoplasmic homeostasis by degrading damaged organelles or misfolded proteins.Citation35-37 Many studies have shown that autophagy is a crucial defense mechanism against a variety of intracellular pathogens, including Shigella flexneri,Citation38 Listeria monocytogenes,Citation39 and M. tb.Citation40 However, our results showed that depletion of GEF-H1 did not affect autophagy in macrophages.
Upon infection, alveolar macrophages phagocytose mycobacteria, but ultimately fail to destroy them completely. Studies have demonstrated that GEF-H1 enhances TNF production in response to bacterial infection,Citation12 and tumor necrosis factor (TNF) in turn elevates macrophage activity to eliminate intracellular bacteria, indicating that GEF-H1 may be required in the macrophage-mediated immune defense against bacterial infection.Citation41-43 In present study, we found that GEF-H1 contributed to the decreased survival of intracellular BCG at all tested time points, indicating that GEF-H1 promoted macrophage-mediated bacterial elimination. Moreover, phagocytosis assays showed that GEF-H1 increased macrophage-mediated mycobacterial phagocytosis.
Collectively, our results demonstrated, for the first time, that GEF-H1 was induced in macrophages upon mycobacterial infection. GEF-H1 promoted cytokine production and mycobacterial elimination in macrophages. Our findings identified GEF-H1 as a novel regulator of cytokine production and mycobacterial elimination, providing important insights into the development of effective therapeutic treatments of TB.
Materials and methods
Ethics statement
All animal experiments in this study were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All experimental protocols were reviewed and approved by the Medical Ethics Board and the Biosafety Management Committee of Southern Medical University.
Antibodies and chemicals
The following reagents were used in this study: TRIzol reagent (15596–018; Invitrogen, Carlsbad, CA, USA), Texas-red (T-6134; Invitrogen), Middlebrook 7H9 Broth (271310; BD Difco Laboratories), and MiddleBrook 7H10 Agar (262710; BD Difco Laboratories). The following antibodies were used in this study: anti-LC3 (NB100–2220; Novus Biologicals), anti-glyceraldehyde 3-phosphate dehydrogenase (TA-08; ZSGB-BIO), anti-β-tubulin (2128; Cell Signaling Technology, Danvers, MA, USA), anti-GEF-H1 (55B6; Cell Signaling Technology), anti-phospho-GEF-H1 (Ser886; E1L6D; Cell Signaling Technology), anti-p38 MAPK (D13E1; Cell Signaling Technology), anti-phospho-p38 MAPK (Thr180/Tyr182; 3D7; Cell Signaling Technology), anti-TBK1/NAK (D1B4; Cell Signaling Technology), anti-phospho-TBK1/NAK (Ser172; D52C2; Cell Signaling Technology), anti-p44/42 MAPK (ERK1/2; 137F5; Cell Signaling Technology), anti-phospho-p44/42 MAPK (ERK1/2; Thr202/Tyr204; D13.14.4E; Cell Signaling Technology), anti-JNK (sc-571; Santa Cruz, Biotechnology, Santa Cruz, CA, USA), and anti-phospho-JNK (sc-6254; Santa Cruz Biotechnology).
Bacterial and cell culture
M. tb H37Rv strain 25618 and M. bovis BCG strain 19015 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), and mycobacteria were grown in Middlebrook 7H9 broth medium or on 7H10 agar plates supplemented with OADC and cultured in a standard tissue culture incubator as described previously.Citation15 RAW 264.7 cells (ATCC; TIB-71) were cultured in in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin.
Isolation and culture of murine BMDMs
Eight-week-old female C57BL/6 mice were purchased from Southern Medical University Animal Supply Center (Guangdong Province, China). For BMDM preparation, single-cell suspensions of bone marrow cells from the femurs and tibiae of mice were cultured in DMEM containing 10% FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin, supplemented with 100 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF). Fresh medium was provided on days 3 and 5 of culture. On day 7 of culture, the isolated macrophages were used for further in vitro studies.
siRNA and transient transfection
Scrambled NC siRNA was purchased from RiboBio Co. RAW264.7 cells (at approximately 50% confluence) were transiently transfected with 40 pM Si-NC or si-GEF-H1 (Si-GEF-H1–1: GGCGACGCTTTATACTTGA; Si-GEF-H1–2: CCAAGGCCTTAAAGCTCTA; Si-GEF-H1–3: GCTCGTGTTTCTCCAGGAA) or 1.6 μg plasmid; using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Western blotting assay
Western blotting was performed as described previously.Citation16 Briefly, whole-cell extracts were resolved by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% bovine serum albumin (BSA) and then incubated with diluted primary antibodies at 4 °C overnight. Western blots were developed using horseradish peroxidase-conjugated secondary antibodies, followed by detection with enhanced chemiluminescence.
Colony-forming unit (CFU) assay
RAW 264.7 cells were infected with BCG at an MOI of 10. After 1 h of incubation at 37 °C, the infected cells were washed extensively with phosphate-buffered saline (PBS) to remove extracellular mycobacteria, incubated for the indicated times, and then lysed in 1 mL distilled water. Quantitative culturing was performed using 10-fold serial dilutions. Aliquots of each dilution were inoculated in triplicate on Middlebrook 7H10 agar plates supplemented with OADC. Plates were incubated for 3 weeks until colonies were counted. The amount of intracellular viable bacilli was determined by the number of colonies calculated. The survival rate of BCG at 24, 48, 72 h were calculated compared with that at 0 h.
ELISA
For RAW264.7 cells and BMDMs experiments, cell-free culture supernatants were collected at 24 h after incubation with BCG (MOI = 2) and used to measure IL-6 (EM004–96; ExCell, Zhejiang Province, China) and IL-1β (EM001–96; ExCell, Zhejiang Province, China) by ELISA according to the manufacturer's instructions.
Griess assay
NO production was determined by measuring its stable end product nitrite, using a Griess reagent (G2930; Promega, Madison, WI, USA) according to the manufacturer's protocol. Briefly, 50 μL of supernatant was added to each well of a 96-well plate, followed by 50 μL sulphanilamide and 50 μL N-1-napthylethylenediamine dihydrochloride (NED). Absorbance at 535 nm was measured using a microplate reader and nitrite concentrations were estimated using a standard nitrite curve. For NO production in RAW264.7 cells, nitrite concentration in the corresponding conditioned medium was subtracted from that in RAW264.7 cells supernatants.
Phagocytosis assays
Phagocytosis was assayed by flow cytometry, as described previously.Citation15 Briefly, BCG was incubated with Texas Red (Invitrogen) at room temperature for 2 h, protected from light, and then gently rinsed with PBS. RAW264.7 cells were then challenged with Texas Red -labeled BCG at an MOI of 10. After 1 h of incubation, cells were washed 3 times with cold PBS, centrifuged to remove extracellular bacteria, and then analyzed by flow cytometry using a Life Science Solution Group instrument (Attune NxT).
RNA isolation and real-time-PCR
Total RNA was isolated with TRIzol reagent, as described previously,Citation16 and 1 μg of RNA was then reverse transcribed into cDNA using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (AT311–03; TransGen Biotech, China). Real-time PCR was performed using TransStart Top Green qPCR SuperMix (TransGen Biotech) on a Mastercycler ep realplex4 (Eppendorf, Hamburg, Germany). The PCR conditions included an initial step at 95 °C for 30 s, followed by 40 cycles of amplification and quantification (95 °C for 5 s, 60 °C for 30 s). β-Actin was used as an internal control. Relative gene expression levels were calculated using the 2−ΔΔCt method.Citation44 The primer sequences used for PCR were as follows: β-Actin, 5′-GATTACTGCTCTGGCTCCTAGC-3′ (forward) and 5′-GACTCATCGTACTCCTGCTTGC-3′ (reverse); IL-1β, 5′-TGTCCTCATCCTGGAAGGTCCACG-3′ (forward) and 5′-CGCAGCAGCACATCAACAAGAGC-3′ (reverse); IL-6, 5′-ACAGAGGATACCACTCCCAACAGA-3′ (forward) and 5′-CTGCAAGTGCATCATCGTTGTTCA-3′ (reverse); IFN-β, 5′-GGAGATGACGGAGAAGATGCAGA-3′ (forward) and 5′-GGAGTTCATCCAGGAGACGTACA-3′ (reverse); GEF-H1, 5′-CAGTCTGTCTCCCTTCGAAGTAAGA-3′ (forward) and 5′-CAGCAATGTTGGTAGTGGAAACACT-3′ (reverse); and iNOS, 5′-TCCTCATGGGACAGCACAGAATG-3′ (forward) and 5′-GTGTCATGCAAAATCTCTCCACTGCC-3′ (reverse).
Statistical analysis
Data are shown as means ± standard errors of the means (SEMs). Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). Differences between 2 groups were compared by using Student's t-test. Differences with p values of less than 0.05 were considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Science and Technology Key Projects on Major Infectious Diseases under Grant number 2017ZX10201301–008; National Natural Science Foundation of China under Grant number 81571951, 81371764; Guangdong Natural Science Foundation under Grant number 2016A030311001, 2014A030313322; and Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme under Grant number 2012.
References
- WHO. Global actions and investments fall far short of those needed to end the global TB epidemic., 2016
- Lin PL, Flynn JL. Understanding latent tuberculosis: a moving target. J Immunol 2010; 185:15-22; PMID:20562268; https://doi.org/10.4049/jimmunol.0903856
- Singh PP, Goyal A. Interleukin-6: a potent biomarker of mycobacterial infection. Springerplus 2013; 2:686; PMID:24455461; https://doi.org/10.1186/2193-1801-2-686
- Guirado E, Schlesinger LS. Modeling the Mycobacterium tuberculosis Granuloma - the Critical Battlefield in host immunity and disease. Front Immunol 2013; 4:98; PMID:23626591; https://doi.org/10.3389/fimmu.2013.00098
- Landes MB, Rajaram MV, Nguyen H, Schlesinger LS. Role for NOD2 in Mycobacterium tuberculosis-induced iNOS expression and NO production in human macrophages. J Leukoc Biol 2015; 97:1111-9; PMID:25801769; https://doi.org/10.1189/jlb.3A1114-557R
- Zhao Y, Alonso C, Ballester I, Song JH, Chang SY, Guleng B, Arihiro S, Murray PJ, Xavier R, Kobayashi KS, et al. Control of NOD2 and Rip2-dependent innate immune activation by GEF-H1. Inflamm Bowel Dis 2012; 18:603-12; PMID:21887730; https://doi.org/10.1002/ibd.21851
- Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 2002; 4:294-301; PMID:11912491; https://doi.org/10.1038/ncb773
- Guo F, Tang J, Zhou Z, Dou Y, Van Lonkhuyzen D, Gao C, Huan J. GEF-H1-RhoA signaling pathway mediates LPS-induced NF-kappaB transactivation and IL-8 synthesis in endothelial cells. Mol Immunol 2012; 50:98-107; PMID:22226472; https://doi.org/10.1016/j.molimm.2011.12.009
- Chiang HS, Zhao Y, Song JH, Liu S, Wang N, Terhorst C, Sharpe AH, Basavappa M, Jeffrey KL, Reinecker HC. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat Immunol 2014; 15:63-71; PMID:24270516; https://doi.org/10.1038/ni.2766
- Meiri D, Marshall CB, Greeve MA, Kim B, Balan M, Suarez F, Bakal C, Wu C, Larose J, Fine N, et al. Mechanistic insight into the microtubule and actin cytoskeleton coupling through dynein-dependent RhoGEF inhibition. Mol Cell 2012; 45:642-55; PMID:22405273; https://doi.org/10.1016/j.molcel.2012.01.027
- Birkenfeld J, Nalbant P, Yoon SH, Bokoch GM. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol 2008; 18:210-9; PMID:18394899; https://doi.org/10.1016/j.tcb.2008.02.006
- Fukazawa A, Alonso C, Kurachi K, Gupta S, Lesser CF, McCormick BA, Reinecker HC. GEF-H1 mediated control of NOD1 dependent NF-kappaB activation by Shigella effectors. PLoS Pathog 2008; 4:e1000228; PMID:19043560; https://doi.org/10.1371/journal.ppat.1000228
- Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol 2015; 36:161-78; PMID:25687683; https://doi.org/10.1016/j.it.2015.01.003
- Yang K, Wu Y, Xie H, Li M, Ming S, Li L, Li M, Wu M, Gong S, Huang X. Macrophage-mediated inflammatory response decreases mycobacterial survival in mouse MSCs by augmenting NO production. Sci Rep 2016; 6:27326; PMID:27251437; https://doi.org/10.1038/srep27326
- Wang J, Yang K, Zhou L, Minhaowu, Wu Y, Zhu M, Lai X, Chen T, Feng L, Li M, et al. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog 2013; 9:e1003697; PMID:24130493; https://doi.org/10.1371/journal.ppat.1003697
- Wang J, Wu M, Wen J, Yang K, Li M, Zhan X, Feng L, Li M, Huang X. MicroRNA-155 induction by Mycobacterium bovis BCG enhances ROS production through targeting SHIP1. Mol Immunol 2014; 62:29-36; PMID:24937178; https://doi.org/10.1016/j.molimm.2014.05.012
- Yu CH, Micaroni M, Puyskens A, Schultz TE, Yeo JC, Stanley AC, Lucas M, Kurihara J, Dobos KM, Stow JL, et al. RP105 engages phosphatidylinositol 3-Kinase p110delta to facilitate the trafficking and secretion of cytokines in macrophages during Mycobacterial infection. J Immunol 2015; 195:3890-900; PMID:26371254; https://doi.org/10.4049/jimmunol.1500017
- Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 2002; 46:709-17; PMID:12410828; https://doi.org/10.1046/j.1365-2958.2002.03237.x
- Malen H, Berven FS, Softeland T, Arntzen MO, D'Santos CS, De Souza GA, Wiker HG. Membrane and membrane-associated proteins in Triton X-114 extracts of Mycobacterium bovis BCG identified using a combination of gel-based and gel-free fractionation strategies. Proteomics 2008; 8:1859-70; PMID:18442171; https://doi.org/10.1002/pmic.200700528
- Wittmann T, Waterman-Storer CM. Cell motility: can Rho GTPases and microtubules point the way? J Cell Sci 2001; 114:3795-803; PMID:11719546
- Pathak R, Dermardirossian C. GEF-H1: orchestrating the interplay between cytoskeleton and vesicle trafficking. Small GTPases 2013; 4:174-9; PMID:23648943; https://doi.org/10.4161/sgtp.24616
- Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature 2002; 416:190-4; PMID:11894097; https://doi.org/10.1038/416190a
- Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, Medzhitov R, Flavell RA. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 2002; 416:194-9; PMID:11894098; https://doi.org/10.1038/416194a
- Kufer TA, Kremmer E, Adam AC, Philpott DJ, Sansonetti PJ. The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol 2008; 10:477-86; PMID:17970764
- Cullis J, Meiri D, Sandi MJ, Radulovich N, Kent OA, Medrano M, Mokady D, Normand J, Larose J, Marcotte R, et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell 2014; 25:181-95; PMID:24525234; https://doi.org/10.1016/j.ccr.2014.01.025
- Liu G, Yang H. Modulation of macrophage activation and programming in immunity. J Cell Physiol 2013; 228:502-12; PMID:22777800; https://doi.org/10.1002/jcp.24157
- Abdul-Aziz M, Tsolaki AG, Kouser L, Carroll MV, Al-Ahdal MN, Sim RB, Kishore U. Complement factor H interferes with Mycobacterium bovis BCG entry into macrophages and modulates the pro-inflammatory cytokine response. Immunobiology 2016; 221:944-52; PMID:27262511; https://doi.org/10.1016/j.imbio.2016.05.011
- Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, et al. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 2010; 184:3326-30; PMID:20200276; https://doi.org/10.4049/jimmunol.0904189
- Leal IS, Smedegard B, Andersen P, Appelberg R. Interleukin-6 and interleukin-12 participate in induction of a type 1 protective T-cell response during vaccination with a tuberculosis subunit vaccine. Infect Immun 1999; 67:5747-54; PMID:10531224
- Kim KH, Yang CS, Shin AR, Jeon SR, Park JK, Kim HJ, Jo EK. Mycobacterial Heparin-binding Hemagglutinin Antigen Activates Inflammatory Responses through PI3-K/Akt, NF-kappaB, and MAPK Pathways. Immune Netw 2011; 11:123-33; PMID:21637390; https://doi.org/10.4110/in.2011.11.2.123
- Pathak SK, Bhattacharyya A, Pathak S, Basak C, Mandal D, Kundu M, Basu J. Toll-like receptor 2 and mitogen- and stress-activated kinase 1 are effectors of Mycobacterium avium-induced cyclooxygenase-2 expression in macrophages. J Biol Chem 2004; 279:55127-36; PMID:15496409; https://doi.org/10.1074/jbc.M409885200
- Yang CS, Lee JS, Jung SB, Oh JH, Song CH, Kim HJ, Park JK, Paik TH, Jo EK. Differential regulation of interleukin-12 and tumour necrosis factor-alpha by phosphatidylinositol 3-kinase and ERK 1/2 pathways during Mycobacterium tuberculosis infection. Clin Exp Immunol 2006; 143:150-60; PMID:16367946; https://doi.org/10.1111/j.1365-2249.2005.02966.x
- Wang T, Lafuse WP, Zwilling BS. NFkappaB and Sp1 elements are necessary for maximal transcription of toll-like receptor 2 induced by Mycobacterium avium. J Immunol 2001; 167:6924-32; PMID:11739511; https://doi.org/10.4049/jimmunol.167.12.6924
- Chan ED, Chan J, Schluger NW. What is the role of nitric oxide in murine and human host defense against tuberculosis?Current knowledge. Am J Respir Cell Mol Biol 2001; 25:606-12; PMID:11713103; https://doi.org/10.1165/ajrcmb.25.5.4487
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; https://doi.org/10.1016/j.cell.2007.12.018
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; https://doi.org/10.1038/nature06639
- Hurley JH, Schulman BA. Atomistic autophagy: the structures of cellular self-digestion. Cell 2014; 157:300-11; PMID:24725401; https://doi.org/10.1016/j.cell.2014.01.070
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science 2005; 307:727-31; PMID:15576571; https://doi.org/10.1126/science.1106036
- Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol 2003; 5:455-68; PMID:12814436; https://doi.org/10.1046/j.1462-5822.2003.00292.x
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753-66; PMID:15607973; https://doi.org/10.1016/j.cell.2004.11.038
- Rajaram MV, Ni B, Morris JD, Brooks MN, Carlson TK, Bakthavachalu B, Schoenberg DR, Torrelles JB, Schlesinger LS. Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. Proc Natl Acad Sci U S A 2011; 108:17408-13; PMID:21969554; https://doi.org/10.1073/pnas.1112660108
- Bermudez LE, Young LS. Tumor necrosis factor, alone or in combination with IL-2, but not IFN-gamma, is associated with macrophage killing of Mycobacterium avium complex. J Immunol 1988; 140:3006-13; PMID:2834450
- Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995; 2:561-72; PMID:7540941; https://doi.org/10.1016/1074-7613(95)90001-2
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID:11846609; https://doi.org/10.1006/meth.2001.1262