
The adhesion of cells to neighboring cells is essential for tissue morphogenesis and homeostasis. Cells bind to one another via surface adhesion receptors, known as cadherins. In epithelia, the primary cadherin adhesion receptor, E-cadherin, binds to E-cadherin on adjacent cells to establish and maintain cell-cell adhesion. In addition, E-cadherin senses external forces induced by fluid flow, stretching, and compaction. To resist the force, E-cadherin stimulates cell stiffening via RhoA-dependent activation of non-muscle myosin II, increased actin polymerization, and growth of the adhesion complex. The increased actin polymerization and growth of the adhesion complex are energetically costly. Yet, how cells derive the energy needed to support remodeling was previously unknown.
Our recent findings reveal how E-cadherin stimulates energy production to support the changes that allow the cell to resist force.Citation1 Specifically, application of shear stress or tensile force to E-cadherin on 2 different epithelial cell lines triggers activation of AMP-dependent kinase (AMPK, ). AMPK is a serine/threonine kinase that phosphorylates substrates to shutdown energy-consuming biosynthetic pathways (such as fatty acid synthesis, protein translation, cholesterol synthesis) and to promote ATP-producing catabolic pathways (such as glycolysis and fatty acid oxidation).
Figure 1. The signal transduction cascade initiated by E-cadherin in response to force. Active AMPK stimulates increased contractility through Abl tyrosine kinase mediated phosphorylation of Y822 vinculin and the subsequent activation of RhoA and myosin II. Active AMPK also signals for an increase in glucose uptake. The glucose is converted into ATP and provides the energy necessary to support the cytoskeletal rearrangements necessary for cell stiffening.
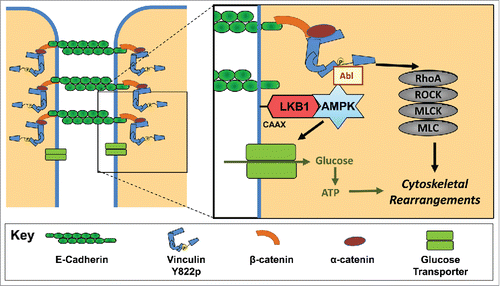
We examined how AMPK is recruited to the cell-cell junctions in response to force. Liver kinase B1, an AMPK activator, localizes to cadherin contacts in mature junctions.Citation2 Application of force increases co-localization and co-immunoprecipitation of LKB1 with E-cadherin.Citation1 When LKB1 is inhibited, force-induced AMPK activation and recruitment to the cadherins is blocked. Hence, LKB1 mediates AMPK recruitment and activation at cell-cell contacts. LKB1 localization to E-cadherin is dependent on a C-terminal CAAX motif.Citation2 Prenylation at similar sites in other proteins promotes their localization to membranes. These observations raise the possibility LKB1 is prenylated in response to force on E-cadherin which triggers LKB1 insertion into the membrane in close proximity to E-cadherin. Alternatively, LKB1 may directly bind to E-cadherin or one of its associated proteins.
The force-induced activation of AMPK raised the possibility that AMPK lies upstream of cellular contractility. E-cadherin triggers increases contractility via a signal transduction cascade whereby Abl kinase phosphorylates Y822 vinculin, thereby stimulating RhoA-dependent myosin II activation.Citation3 Inhibition of LKB1 or AMPK prevents force-induced Abl activation, vinculin phosphorylation, GTP-loading of RhoA, and myosin II phosphorylationCitation1 (). Hence, AMPK is a component of a known signal transduction cascade from E-cadherin to cell stiffening.
In many cells, AMPK signals for increased glucose uptake. In response to force on E-cadherin, we observe increased glucose uptake in 2 cell lines in response to multiple forces.Citation1 The glucose uptake requires increased contractility and E-cadherin expression. Also of note, the glucose taken up into the cell was converted to ATP. Hence, force-induced AMPK activation signals for increased energy within the cell. Interestingly, cells signal for increased energy using the same pathway whereby they stimulate the physical changes necessary to resist the force.
The critical question remains, “Does this increased energy support the physical changes necessary to resist the force?” In response to force, E-cadherin triggers increases in enzymatic activity, actin polymerization, and actomyosin contractility to support cell stiffening. The amount of energy required for these processes can be quite high. Estimates suggest that as much as 50% of the total ATP consumed in a resting platelet or neuron is used to support the actin cytoskeleton.Citation4,5 Our studies reveal a 2.0-fold increase in E-cadherin deposition in cell-cell junctions and a 3.8-fold increase in junctional actin.Citation1 Cells require both AMPK and increased ATP production to support the cytoskeletal changes necessary for cell stiffening. Hence, not only does force-induced AMPK stimulate glucose uptake and increased contractility, but also the energy is used to support elevated tension.
Overall, these studies reveal a complex network of interactions linking E-cadherin mechanotransduction and cell metabolism (). Intriguingly, the newly identified signal transduction cascade provides insight into a poorly understood aspect of cell biology and opens the door to many exciting questions linking cellular mechanics to metabolism. Key among these is how widespread is this phenomenon. AMPK regulates mitochondria trafficking to the leading edge during cell migration and matrix invasion, suggesting that the integrins, like cadherins, may be coupled to metabolic events.Citation6 Another intriguing question arising from this work is whether other metabolic intermediates are linked to cell adhesion. Interestingly, an RNAi screen uncovered 52 metabolic proteins that regulate cell-cell adhesion.Citation7 Hence, it is unlikely AMPK is only metabolic intermediate linked to cadherin-mediated adhesion and mechanotransduction. Identifying these additional links and determining how they regulate cadherin mechanotransduction are now open for investigation.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Bays JL, Campbell HK, Heidema C, Sebbagh M, DeMali KA. Linking E-cadherin mechanotransduction to cell metabolism through force-mediated activation of AMPK. Nature cell biology. 2017;19:724-31. doi:10.1038/ncb3537. PMID:28553939.
- Sebbagh M, Santoni MJ, Hall B, Borg JP, Schwartz MA. Regulation of LKB1/STRAD localization and function by E-cadherin. Curr Biol: CB. 2009;19:37-42. doi:10.1016/j.cub.2008.11.033. PMID:19110428.
- Bays JL, Peng X, Tolbert CE, Guilluy C, Angell AE, Pan Y, Superfine R, Burridge K, DeMali KA. Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions. J Cell Biol. 2014;205:251-63. doi:10.1083/jcb.201309092. PMID:24751539.
- Daniel JL, Molish IR, Robkin L, Holmsen H. Nucleotide exchange between cytosolic ATP and F-actin-bound ADP may be a major energy-utilizing process in unstimulated platelets. European journal of biochemistry / FEBS. 1986;156:677-84. doi:10.1111/j.1432-1033.1986.tb09631.x
- Bernstein BW, Bamburg JR. Actin-ATP hydrolysis is a major energy drain for neurons. J Neurosci. 2003;23:1-6. PMID:12514193.
- Cunniff B, McKenzie AJ, Heintz NH, Howe AK. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell. 2016;27:2662-74. doi:10.1091/mbc.E16-05-0286. PMID:27385336.
- Toret CP, D'Ambrosio MV, Vale RD, Simon MA, Nelson WJ. A genome-wide screen identifies conserved protein hubs required for cadherin-mediated cell-cell adhesion. J Cell Biol. 2014;204:265-79. doi:10.1083/jcb.201306082. PMID:24446484.