
ABSTRACT
Autophagy is a highly conserved process that acts sequestering cytoplasmic components for their degradation by the lysosomes. It consists of several sequential steps that have to be finely regulated to ensure both its progression and termination. Post-translational modifications (PTMs) play an important role in regulating ATG proteins function in different stages of autophagy. Recently, we demonstrated that, during prolonged starvation, ULK1 protein is specifically ubiquitylated by NEDD4L, and that this regulation is important to protect cells against excessive autophagy. In this Extra view, we show that ULK1 phosphorylation at 3 different sites on the same ULK1 target region for NEDD4L is preparatory for its ubiquitylation and subsequent degradation. This adds to the complexity of ULK1 multi-level regulation by several factors, including kinases, phosphatases and acetylases, with each contributing to autophagy homeostasis
Introduction
Protein kinases are important regulators of intracellular processes, and their activity is tightly controlled to induce downstream signals and mediate their physiologic responses. In most cases, kinase activation occurs through phosphorylation on their catalytic domain that results in a conformational change that allows phosphate transfer from ATP to a given substrate.
Interestingly, the activated conformation of a protein kinase is a prerequisite for initiating degradation.Citation1 For the precise control of cellular functions, indeed, many Ser/Thr- and Tyr-specific protein kinases should be degraded and, in the majority of the cases, this occurs through the Ubiquitin Proteasome System (UPS).Citation2 The latter acts as a termination signal that is necessary to regulate and maintain physiologic cellular functions, including cell growth, differentiation, adhesion, motility and death.
Autophagy is a central cellular mechanism for the removal of damaged organelles and protein complexes. This process takes place at a constitutive basal level, even though it can be upregulated in response to various types of stress through very fast and regulated kinetics.Citation3 Its fast response is possible especially through post-translational modifications (PTMs), such as phosphorylation and ubiquitylation of ATG (autophagy-related) proteins.Citation4 The ULK1 (unc-51 like autophagy activating kinase 1; yeast Atg1) kinase is a key player in the early steps of autophagy. Indeed, phosphorylation events play vital roles in autophagy initiation by regulating ULK1 complex assembly and activity. In basal conditions, mTOR acutely inhibits ULK1 functions through phosphorylation.Citation5,6 During nutrient deprivation, instead, ULK1 receives positive inputs from the cellular energy sensor AMP-activated protein kinase (AMPK), this leading to ULK1 activation.Citation7 In addition, it is now evident that, in persistent stress conditions, autophagy is self-inhibited, this avoiding prolonged autophagy to become be detrimental for cell survival.Citation8-11 The main mechanism determining the duration of the autophagy response is the proteasomal degradation of some key autophagy proteins, occurring shortly after their activation. In our recent work,Citation11 we discovered a tight coupling of ULK1 catalytic activity to its degradation. In particular, we found that ULK1 autophosphorylation potentiates its interaction with the E3 ligase NEDD4L that mediates, in turn, its ubiquitylation and leads to ULK1 proteasomal degradation. Indeed, downregulation of NEDD4L leads to a persistent autophagic response, underlining the importance of post-translational modifications in dynamically controlling autophagy.
Crosstalk between phosphorylation and ubiquitylation on ULK1 protein
Phosphorylation and ubiquitylation are 2 of the most prevalent PTMs in eukaryotes that crosstalk at different levels.Citation12,13 In fact, many proteins are multiply modified and many examples are emerging about PTMs combinatorial activity on the same molecule, this adding specificity and peculiarity to signal processing. This crosstalk can be either positive or negative. On the one hand, phosphorylation can promote or inhibit ubiquitylation, which in turn can lead to proteasomal degradation. On the other hand, PTMs can compete for a single residue, by masking the recognition site for a second PTM. According to our recent work,Citation11 we found that ULK1 is specifically downregulated during prolonged starvation by the proteasome. As shown in , indeed, we found that ULK1 protein levels are reduced during the first 4 h of starvation and then restored following a 6 hour starvation (autophagy induction was shown by LC3 lipidation and p62 degradation). Moreover, in our previous publication,Citation11 we identified 2 specific lysine residues on ULK1 that are ubiquitylated by NEDD4L to trigger ULK1 degradation: Lys925 and Lys933. In that work, we demonstrated that mutation of these sites affect both stability and NEDD4L-dependent degradation of ULK1 during autophagy. In our mass spectrometry analysis, performed to identify ULK1-ubiquitylated sitesCitation11 we found that the same peptide carrying 2 modified lysines is also phosphorylated on 2 serine and one threonine residues located at aa 929–931 ().
Figure 1. Phosphorylation of a 3-residues small region (Ser929-Ser930-Thr931) of ULK1 is necessary for its NEDD4L-dependent degradation. (A) HeLa cells were treated with EBSS for the indicated time periods, and levels of ULK1, ACTIN, p62 and LC3 were detected by WB. (B) HeLa cells were transfected with constructs encoding ULK1WT, ULK1AAA and ULK1EDD Myc- tagged proteins (whose sequence is detailed in the top panel) and treated with CHX (50 μM) for different time periods. Levels of ULK1 constructs were detected by WB by using an anti-Myc antibody. Densitometric analysis of ULK1 over ACTIN bands is also shown. (C) HeLa cells were transfected ULK1WT, ULK1AAA, ULK1EDD Myc- tagged proteins together with NEDD4L-HA in the presence or not of MG132 (4h) and in the presence or not of EBSS (6 h). Levels of ULK1, NEDD4L and ACTIN were detected by WB. Densitometric analysis of ULK1 over ACTIN bands is also shown. (D) HeLa cells were transfected as in (B) and autophagy was induced with EBSS for different time periods. The levels of ULK1 and ACTIN were detected by WB. Densitometric analysis of ULK1 over ACTIN bands is also shown. For all ULK1 mutant constructs, the same amount of DNA was used for transfection; the differences are caused by the different stability of these mutants. In (B–D), data are expressed as the mean value ± SEM (n = 3), and were analyzed by one-way ANOVA followed by Turkey post hoc test. *p < 0.05. (E) HeLa cells were transfected with a vector encoding a 6xHIS-tag Ubiquitin together with ULK1WT and ULK1AAA in the presence or not of NEDD4L. Samples were treated with MG132 for 4h before harvest. Protein extracts were prepared in a denaturing urea buffer and subjected to Ni-NTA purification. The amount of ubiquitylated ULK1 co-purified with 6xHIS-Ubiquitin was evaluated by WB. For all ULK1 mutant constructs, the same amount of DNA is used for the transfection: differences are due to the increased stability of these mutants.
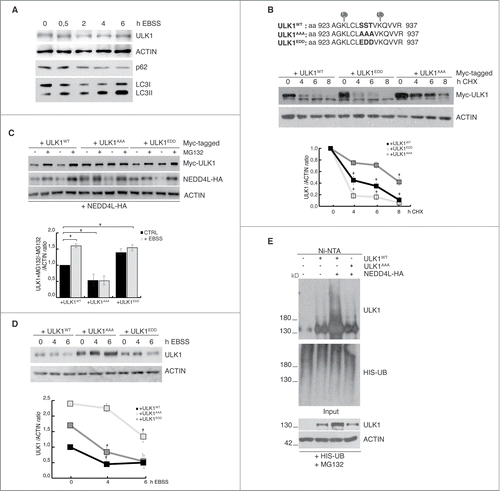
We thus decided to investigate whether these sites could be important for ULK1 stability and ubiquitylation. By site-directed mutagenesis, we generated 2 different mutant constructs: a phospho-silencing mutant (ULK1AAA) replacing the 3 residues (2 serine and one threonine) with alanines, and a phospho-mimicking construct (ULK1EDD) replacing them with one glutamic acid and 2 aspartic acid respectively.
First, we have analyzed the half-life of both mutant constructs in the presence of cycloheximide (CHX), an inhibitor of protein translation. As shown in , we found that upon CHX treatment in a given time-course (0–8 hours), ULK1AAA is more stable than both ULK1WT and ULK1EED; this indicates that the phosphorylation of these sites is important for ULK1 stability. Then, we evaluated the capability of both mutant constructs to be degraded by NEDD4L. Since we have learned that overexpression of wild-type NEDD4L efficiently promotes ULK1 protein decrease,Citation11 we co-expressed all the constructs with NEDD4L in the presence or not of the proteasomal inhibitor MG132 in both basal conditions and upon autophagy induction by starvation. As shown in , the phospho-silencing mutant (ULK1AAA) is the only one exhibiting a negative effect on the NEDD4L-dependent degradation of ULK1.
Based on the evidence that NEDD4L mediates ULK1 degradation during prolonged autophagy,Citation11 we decided to explore whether these phosphorylations affected or not ULK1 stability during starvation. To this aim, we overexpressed these constructs and induced autophagy by starvation for different time periods (0, 4 and 6 hours). Also in these experimental conditions (), the negative effect on ULK1 degradation is restricted to the ULK1AAA mutant construct, this confirming the hypothesis that phosphorylation of these sites is necessary for ULK1 NEDD4L-dependent degradation during autophagy. Finally, we analyzed the capability of NEDD4L to ubiquitylate ULK1AAA by means of an in vivo ubiquitylation assay. As shown in , there is a strong decrease in the ubiquitylation status of the mutant construct when compared with the wt.
Altogether, these experiments support the idea that the simultaneous mutation of these 3 sites (Ser929-Ser930-Thr931) is sufficient to alter ULK1 stability and subsequent NEDD4L-dependent degradation (). These findings open new questions. What is/are the kinase(s) responsible for these phosphorylations? It is a well-known general mechanism that autophosphorylation of a kinase can create binding sites for an E3 ligaseCitation2 or can result in an altered conformation that can be recognized by an E3 ligase to ensure a rapid termination of its activity. In our case, we know that ULK1 autophosphorylation (Ser1047) potentiates its interaction with NEDD4L, this leading to its proteasomal degradation; indeed, the inactive form of ULK1 fails to be degraded during autophagy.Citation11 We can thus speculate that ULK1 itself could be also the responsible kinase for one or more phosphorylations on this small region; it should be noted that these modified sites are located in the C-terminal domain of ULK1 that is commonly responsible for the dynamic binding to its interactors, such as ATG136 and AMBRA1.Citation14 A conformational change or the dissociation of ULK1-interacting proteins could, in principle, expose a degron that would otherwise be masked. Alternatively, transphosphorylation by a different protein kinase that is activated during autophagy could create a phosphodegron that is recognized by a phospho-dependent ligase, such as NEDD4L. In this case, we can hypothesize the activation of a stress-responsive kinase, downstream of mTOR, to inhibit autophagy by promoting ULK1 degradation. However, since we mutated the 3 sites simultaneously we cannot exclude 1) the involvement of more kinases in mediating these phosphorylations and 2) that all the 3 sites are required for NEDD4L-dependent ULK1 ubiquitylation.
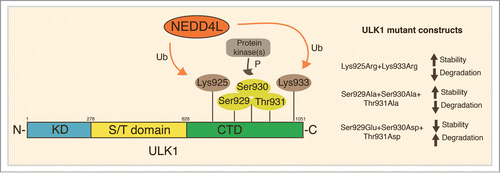
Figure 2. ULK1-modified sites important for its proteasomal degradation. During autophagy induction, the E3 ligase NEDD4L ubiquitylates ULK1 on both Lys925 and Lys933. These modifications regulate ULK1 stability and are necessary for its proteasome degradation. Moreover, phosphorylation of a 3-residues small region (Ser929-Ser930-Thr931) by unknown protein kinase(s) in the same region of ULK1 is also necessary for its ubiquitylation. The simultaneous mutation of these sites is, indeed, sufficient to alter ULK1 stability and subsequent degradation. Ub, Ubiquitylation; P, phosphorylation; KD, kinase domain; S/T, serine/threonine domain; CTD, C-terminal domain.
A disruption of ubiquitylation regulating protein kinase is directly or indirectly linked to human diseases, including cancer. The full understanding of post-translational modifications of key regulators of autophagy (the autophagy modifome) is thus important, not only due to their role in normal cellular functions, but also due to their drug-targeting potential in diseases. Currently, indeed, PTMs are tracked as disease markers or used as molecular targets for developing target-specific therapies, with the establishment of their biologic influence that has become a critical step in early stages of biopharmaceutical drug development.
Materials and methods
Cell culture and reagents
HeLa cells were cultured and transfected as described previously.Citation11
The induction of autophagy by nutrient starvation was obtained by washing cells with PBS and incubating them with Earle's Balanced Salt Solution (EBSS, Sigma Aldrich). Proteasome activity was inhibited by 5 μM MG132 (Sigma-Aldrich); translation was inhibited by 50 μM Cycloheximide (CHX).
Ni-Nta assay
For detection of ULK1 ubiquitylation, Ni-Nta assay was performed as described in our previous publication.Citation11
Plasmids
ULK1 mutant constructs were generated by using the site-directed mutagenesis kit (Agilent Technologies). The sequences used are as follows:
ULK1 AAA: 5′gcaagctctgcctggcggcggccgctgtgaagcaggtggtgcgcag 3′
ULK1 EDD: 5′gcaagctctgcctggaggacgatgtgaagcaggtggtgcgca 3′
Western blot
Cell lysate preparation and immunoblotting were performed as described in Nazio et al. (2016).Citation11 Also, the catalog numbers and dilutions of the shown antibodies were already reported there.Citation11
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank M. Acuña Villa for secretarial work and G. Pericoli for technical assistance.
Funding
Francesco Cecconi's laboratory is supported by grants from Fondazione Roma, AIRC (IG2016–18906), FISM-Fondazione Italiana Sclerosi Multipla -Cod. 2013/R/7 (2013), KBVU from the Danish Cancer Society (R146-A9364), the Novo Nordisk Foundation (7559, 22544) and the European Union (Horizon 2020 MEL-PLEX, grant agreement 642295). Further, FC laboratory in Copenhagen is part of the Center of Excellence in Autophagy, Recycling and Disease (CARD), funded by the Danish National Research Foundation.
References
- Kang BS, French OG, Sando JJ, Hahn CS. Activation-dependent degradation of protein kinase C eta. Oncogene. 2000;19:4263-72. doi:10.1038/sj.onc.1203779. PMID:10980600
- Lu Z, Hunter T. Degradation of activated protein kinases by ubiquitylation. Annu Rev Biochem. 2009;78:435-75. doi:10.1146/annurev.biochem.013008.092711. PMID:19489726
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280-93. doi:10.1016/j.molcel.2010.09.023. PMID:20965422
- Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25(6):354-63. doi:10.1016/j.tcb.2015.02.002. PMID:25759175
- Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy. 2013;9(2):124-37. doi:10.4161/auto.23323. PMID:23295650
- Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009;2(84):pe51. doi:10.1126/scisignal.284pe51. PMID:19690328
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132-41. doi:10.1038/ncb2152. PMID:21258367
- Antonioli M, Albiero F, Nazio F, Vescovo T, Perdomo AB, Corazzari M, Marsella C, Piselli P, Gretzmeier C, Dengjel J, et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamic. Dev Cell. 2014;31(6):734-46. doi:10.1016/j.devcel.2014.11.013. PMID:25499913
- Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF, et al. Cul3-KLHL20 Ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell. 2016;61(1):84-97. doi:10.1016/j.molcel.2015.11.001. PMID:26687681
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942-6. doi:10.1038/nature09076. PMID:20526321
- Nazio F, Carinci M, Valacca C, Bielli P, Strappazzon F, Antonioli M, Ciccosanti F, Rodolfo C, Campello S, Fimia GM, et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J Cell Biol. 2016;215(6):841-56. doi:10.1083/jcb.201605089. PMID:27932573
- Hunter T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28(5):730-8. doi:10.1016/j.molcel.2007.11.019. PMID:18082598
- Nguyen LK, Kolch W, Kholodenko BN. When ubiquitination meets phosphorylation: A systems biology perspective of EGFR/MAPK signalling. Cell Commun Signal. 2013;11:52. doi:10.1186/1478-811X-11-52. PMID:23902637
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406-16. doi:10.1038/ncb2708. PMID:23524951