
ABSTRACT
The tumor suppressor protein p53 interacts with DNA in a sequence-dependent manner. Thousands of p53 binding sites have been mapped genome-wide in normal and cancer cells. However, the way p53 selectively binds its cognate sites in different types of cells is not fully understood. Here, we performed a comprehensive analysis of 25 published p53 cistromes and identified 3,551 and 6,039 ‘high-confidence’ binding sites in normal and cancer cells, respectively. Our analysis revealed 2 distinct epigenetic features underlying p53-DNA interactions in vivo. First, p53 binding sites are associated with transcriptionally active histone marks (H3K4me3 and H3K36me3) in normal-cell chromatin, but with repressive histone marks (H3K27me3) in cancer-cell chromatin. Second, p53 binding sites in cancer cells are characterized by a lower level of DNA methylation than their counterparts in normal cells, probably related to global hypomethylation in cancers. Intriguingly, regardless of the cell type, p53 sites are highly enriched in the endogenous retroviral elements of the ERV1 family, highlighting the importance of this repeat family in shaping the transcriptional network of p53. Moreover, the p53 sites exhibit an unusual combination of chromatin patterns: high nucleosome occupancy and, at the same time, high sensitivity to DNase I. Our results suggest that p53 can access its target sites in a chromatin environment that is non-permissive to most DNA-binding transcription factors, which may allow p53 to act as a pioneer transcription factor in the context of chromatin.
Introduction
The tumor suppressor p53 is a DNA-binding transcription factor (TF) that plays a pivotal role in preventing cancer growth.Citation1 p53 interacts with target DNA sites in a sequence-dependent manner, with the consensus motifs comprising 2 decanucleotides RRRCWWGYYY (R = A, G; Y = C, T; W = A, T), separated by a variable spacer.Citation2,3 Two approaches have been used to map p53 binding sites. The first approach is a traditional, single-gene method that usually uses reporter-gene constructs to determine the specific DNA fragments interacting with p53 in vitro,Citation3 and these p53 sites are referred to as response elements (REs). The second approach is based on chromatin immune-precipitation (ChIP), an experimental technique used to investigate protein-DNA interactions in vivoCitation4; these p53 sites are denoted as ChIP fragments. p53 REs and ChIP fragments are distributed differently in the human genome. Compared to REs, most p53 ChIP fragments are found far from transcriptional start sites (TSSs),Citation5 and some of them reside in interspersed repeats such as retroviral terminal repeats (LTRs),Citation6 short interspersed nuclear elements (SINEs),Citation7,Citation8 and long interspersed nuclear elements (LINEs).Citation9 Interestingly, quite a few p53 REsCitation7 and ChIP fragmentsCitation8 reside in primate-specific Alu repeats, suggesting that Alu repeats play an important role in shaping the p53 regulatory network in the context of chromatin.
p53-DNA binding in vivo is influenced by local chromatin states.Citation10 The first evidence of this was reported in the seminal study by Espinosa and Emerson, who assessed the relative affinities of p53 for its 2 target sites within the CDKN1A/p21 promoter and found that p53 exhibits a higher affinity to the sites in the context of chromatin than in naked DNA.Citation11 Moreover, p53-dependent changes in histone acetylation have been observed at the promoters of many p53 target genes, including CDKN1A/p2112 and MDM213. H3 and H4 histone modifications have been observed for 20% of target genes after p53 is overexpressed.Citation14,15 Recent studies revealed that the transcriptionally active histone mark, histone H3 lysine 4 trimethylation (H3K4me3), is associated with both p53 binding and transactivation of target genes,Citation16 and the repressive histone mark, H3K27me3, is detected in repressed p53 target genes.Citation17 All these observations suggest that p53 binding coordinates with histone modifications to regulate gene transcription. The level of DNA methylation at p53 binding sites was also shown to be critical for in vivo DNA binding.Citation18,19
TF-DNA binding in vivo is usually associated with open chromatin, which is characterized by increased DNase I sensitivity,Citation20 and an integrative analysis of ChIP fragments bound by 119 human TFs revealed that most TF binding sites are located in nucleosome-depleted DNase I sensitive regions.Citation21 However, another group of proteins (including p53) known as pioneer factors are able to interact with nucleosomal DNA.Citation22-24 Indeed, it has been shown that p53 can interact with nucleosomal DNA both in vitro,Citation25,Citation26 and in vivo.Citation26,27 This finding is consistent with previous studies showing that well-known p53 REs including the p21 5′ RE are often exposed on the nucleosomal surfaceCitation28 and occur in chromatin domains that are resistant to DNase I digestion.Citation29 Recent studies however have showed that the binding sites of 2 pioneer factors, progesterone receptor (PR)Citation30 and forehead box protein A2 (Foxa2),Citation31 occur in genomic regions with both high nucleosome occupancy and high sensitivity to DNase I, suggesting that high sensitivity to DNase I does not necessarily reflect nucleosome depletion. Whether p53 in vivo binding sites also occur in genomic regions displaying high DNase I sensitivity remains unknown.
All these studies suggest that p53-DNA binding in vivo is associated with multiple chromatin patterns (i.e., histone modifications, DNA methylation, nucleosome occupancy and DNase I sensitivity). Because normal cells (NCs) and cancer cells (CCs) are characterized by drastically different chromatin organization,Citation32,33 we hypothesize that p53 binding sites derived from the 2 different types of cells exhibit distinct epigenetic patterns.
In the present study, we conducted a comprehensive analysis of 25 published p53 cistromes that include more than 120,000 p53 ChIP fragments from numerous NC and CC lines under various stress conditions. Merging these data sets produces 6,039 NC and 3,551 CC ‘high-confidence’ p53 ChIP clusters that include 3 or more overlapping ChIP fragments. These clusters are enriched in the endogenous retrovirus 1 (ERV1) repeat family of the human genome. We found that regardless of the cell type, the p53 ChIP clusters occur in genomic regions with high nucleosome occupancy and high DNase I sensitivity, which differs from most DNA-binding TFs. Finally, compared with their counterparts in cancer cells, the NC ChIP clusters are more likely associated with a higher level of transcriptionally active histone marks (H3K4me3 and H3K36me3) and DNA methylation, but a lower level of repressive histone marks (H3K27me3). In light of these findings, a new scheme was proposed to explain the distinct p53 genomic binding patterns in normal and cancer cells.
Results
Comparison between p53 binding sites identified in vitro and in vivo
To compare the p53 binding sites identified in vitro and in vivo, we collected 154 p53 REs (Table S1) and ∼120,000 p53 ChIP fragments from 25 published data sets, which include 42,020 NC ChIP fragments in 9 data sets and 77,796 CC ChIP fragments in 16 data sets (, Table S2).
Figure 1. Analysis of p53 ChIP clusters derived from human normal and cancer cells. (A) Schematic view of ChIP fragment analysis. Nine data sets from normal cell (NC) lines and 16 data sets from cancer cell (CC) lines were used in this study (Table S2). The positions of ChIP fragments were converted to human genome assembly hg18 using the LiftOver utility in the UCSC Genome Browser. Overlapping ChIP fragments form clusters. A pile-3+ cluster containing 3 or more overlapping members is denoted as a ChIP cluster and used for further analysis. Arrows denote the lengths of clusters. (B) Length distribution of ChIP clusters from NC (left) and CC (right) data sets. The x-axis represents ChIP cluster lengths binned into 100-bp intervals. The y-axis represents the occurrence of ChIP clusters in each bin. (C) Overlap of ChIP clusters from NCs and CCs. The p53 ChIP clusters are naturally divided into 3 groups: NC-only, N/C, and CC-only.
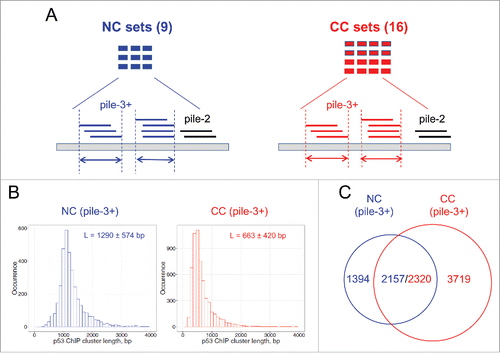
The NC ChIP fragments are significantly longer than those from CC lines (Wilcoxon test, p < 2.2 × 10−16; Fig. S1). Note that the longest p53 ChIP fragments came from human embryonic stem cells (hESCs)Citation34 and keratinocytesCitation35 (Fig. S2). By contrast, the CC ChIP fragments have a narrower length distribution, with an average length of approximately 500 bp (Fig. S3). Although the possibility of experimental variations cannot be ruled out, this difference in ChIP fragment lengths could be due to distinct chromatin structures in NCs and CCs. That is, cancer chromatin may have a more ‘extended’ structure due to global hypomethylation,Citation32,33 which would facilitate chromatin shearing in ChIP-seq experiments. We note that the observed difference in ChIP fragment lengths between NC and CC groups is unlikely to have any effect on the following analysis.
To investigate how p53 REs identified in vitro overlap with p53 ChIP fragments obtained in vivo, we visualized p53 ChIP fragments in known target genes using the UCSC Genome Browser (Fig. S4). We found that out of 154 p53 REs, 71 REs (46%) overlap with at least one NC or CC ChIP fragment (Table S1).
For a detailed comparison, we selected 2 well studied p53 target genes, CDKN1A/p2136 and PUMA.Citation37 We found that multiple ChIP fragments contain previously reported p53 REs in these 2 genes (Figs. S5 and S6), demonstrating a good agreement between p53 REs and ChIP fragments. Analysis of ChIP fragments in these 2 genes led to the following observations.
First, normal and cancer cells differ in p53 ChIP occupancy at previously reported p53 REs. Consistent p53 occupancy is often observed in cancer but not in normal cell lines. For example, 14 out of 16 (88%) CC ChIP data sets show p53 occupancy at the 5′ or 3′ REs in the p21 promoter, whereas only 4 out of 9 (44%) NC data sets have signals at these positions (Fig. S5). Likewise, 12 out of 16 (75%) CC data sets have p53 occupancy at the known PUMA RE, whereas only 2 out of 9 (22%) NC data sets show signals around the same position (Fig. S6). Further analysis of NC data sets, for example, the ChIP fragments from keratinocytes,Citation35 showed that p53 occupancy at the p21 and PUMA REs occur in both untreated (control) and treated conditions. Because only drug-induced p53 ChIP fragments were analyzed in our study (see Methods), these ChIP fragments are not shown in Figure S5 and S6.
Second, in addition to known p53 REs, ChIP fragments overlap at other locations, which may have functional significance. For example, in the PUMA gene, 5 of 9 (56%) of NC data sets have p53 occupancy ∼2 kb downstream of the end of PUMA gene, while only 2 of 16 (13%) of CC data sets reveal detectable occupancy of this binding site, suggesting that it is likely to be NC-specific. On the other hand, in the p21 gene, multiple ChIP fragments are located at an intronic locus, ∼3.3 kb downstream of TSS (black arrow in Fig. S5). This genomic locus corresponds to a previously identified p53 binding site that can activate a variant of the p21 gene, p21B, to induce apoptosis.Citation38 This result suggests that superimposition of multiple ChIP fragments from different data sets helps identify functionally important p53 binding sites. Overall, we observed distinctive p53 binding patterns in normal and cancer cells under drug treatments, in agreement with earlier studies.Citation18,19
Searching for high-confidence p53 binding sites in normal and cancer cells
We set out to assemble the NC and CC ChIP fragments into clusters across the human genome (hg18). About 40% of the ChIP fragments are singletons that are scattered throughout the genome, whereas the remaining fragments overlap with each other and form clusters ( and Table S3). The clusters are denoted pile-2, pile-3, pile-4, and so forth based on their coverage (i.e., number of overlapping members in a cluster). Our Monte Carlo simulation analysis showed that pile-3+ clusters are highly unlikely to form by chance (p < 0.01, see Methods and Table S3), which suggests that these clusters reflect ‘real’ ChIP enrichment in normal and cancer cells.
Thus, we identified 3,551 NC and 6,039 CC pile-3+ clusters (for brevity, denoted NC and CC ChIP clusters, respectively). We found that the lengths of NC and CC ChIP clusters differ significantly (Wilcoxon test, p < 2.2 × 10−16, ). We also found that 2,157 (61%) of the NC ChIP clusters overlap with the CC ChIP clusters, whereas 2,320 (38%) of the CC ChIP clusters overlap with the NC ChIP clusters (, Tables S4-S5). This small discrepancy in the number of overlapping clusters (2,157 vs. 2,320) is likely due to the different length distributions in NC and CC ChIP clusters (). In other words, a typical NC ChIP cluster (∼1,300 bp in length) may cover 2 CC clusters (∼700 bp in length). The 2,320 ChIP clusters from CCs were included in the “normal/cancer” (i.e., N/C) group for the studies described below (because they have a higher number than their counterparts from normal cells). The ChIP clusters that are not shared by the normal and cancer data sets are put into the “CC-only” group or the “NC-only” group, respectively.
Noticeably, most p53 ChIP clusters occur more than 10 kb away from the TSSs of the nearest genes, which is in sharp contrast to p53 REs (Chi-squared test p < 6.9 × 10−7, Fig. S7). This result indicates that p53 binding sites identified in vitro and in vivo exhibit distinctive genomic distributions, consistent with earlier studies.Citation5,7
Gene ontology analysis by DAVIDCitation39,40 revealed that ChIP clusters are enriched in genes involved in p53 signaling pathways. Consider the N/C group as an example. The most significant KEGG pathway is the “p53 signaling pathway,” which has an enrichment score of 9.6 (Fisher's exact test, p < 0.01) (Fig. S8A). For this reason, most of enriched gene ontology terms are related to p53 functions, including “regulation of apoptosis,” “DNA damage response,” and “regulation of cell cycle” (Fig. S8B).
Distribution of p53 motifs in high-confidence binding sites in human repetitive elements
To examine the relationship between p53 ChIP clusters and repetitive elements in the human genome, we first identified p53 motifs (∼20 bp in length) in each cluster using our previously developed computational methodCitation7 based on a position weight matrix (PWM) formalism (). This method scores DNA fragments based on their similarity to the consensus p53 binding motif. That is, the more similar to the consensus sequence, the higher the PWM score. A cutoff of 70% suggested in our previous studyCitation7 was used. If multiple p53 motifs with the score 70% or higher are identified in a given p53 ChIP cluster, the motif with the highest score is selected. As a result, 3,298 (93%) motifs were identified in 3,551 NC ChIP clusters, whereas 4,791 (76%) motifs were found in 6,309 CC ChIP clusters (Tables S4-S5). Note that a substantial fraction of p53 ChIP clusters (∼7% NC and ∼24% CC clusters) contains no obvious p53 binding motifs; a similar trend was also observed in other studies.Citation5,41
Figure 2. Analysis of p53 motifs in ChIP clusters. (A) Overall research plan for the p53 motifs identified in ChIP clusters. The PWM-based tool developed in our previous study (Cui et al. Citation2011) was used to predict p53 motifs. If multiple p53 motifs were found in a given cluster, the motif with the highest PWM score was selected. (B) Locations of p53 motifs relative to the ChIP cluster centers. (C) Distribution of p53 motif scores with respect to the coverage of ChIP clusters.
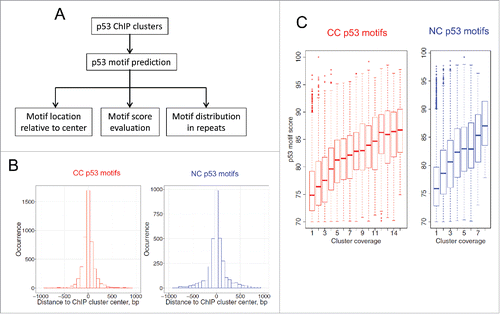
There are several possible reasons for the lack of p53 binding motifs in these ChIP clusters. First, in certain cases, p53 binds DNA based on the topology of a DNA fragment, not on its sequence.Citation42 Second, p53 can bind to different motifs such as microsatellites in the PIG3 gene.Citation43 Third, p53 can be recruited to target sites through indirect DNA binding, as shown in the p53-dependent transcriptional repression of the survivin gene.Citation44
We found that the identified p53 motifs tend to occur close to the centers of p53 ChIP clusters for both NC and CC data sets (), suggesting that the length differences observed in have no effect on our analysis of p53 binding motifs. Moreover, the average motif scores increase with the coverage of p53 clusters (), indicating that the clusters with more overlapping members tend to have stronger p53 motifs. To check whether the p53 motifs overlap with repeat elements, we compared the locations of motifs and the RepeatMasker annotation of the human genome (hg18). We found that about 40% of the p53 motifs overlap with various types of repetitive elements, including LINE, SINE, LTR and simple repeats (), consistent with earlier studies.Citation6-9 Analysis of the repeat-associated p53 motifs yielded the following observations.
Figure 3. Distribution of identified p53 motifs in the human genome (A–C) and in the ERV/LTR repeat family (D–F). ChIP clusters from the NC-only group (A, D), the N/C group (B, E), and the CC-only group (C, F) were taken into consideration. RepeatMasker annotation of the human genome (hg18) downloaded from the UCSC Genome Browser was used for analysis. If a given p53 motif does not overlap with any repetitive element in the genome, it was designated as “Non_repeat.” Otherwise, the motif was assigned to the corresponding repeat family and subfamily. The fractions of p53 motifs in repeat (sub)families and in the “Non_repeat” group were calculated with respect to the coverage of ChIP clusters.
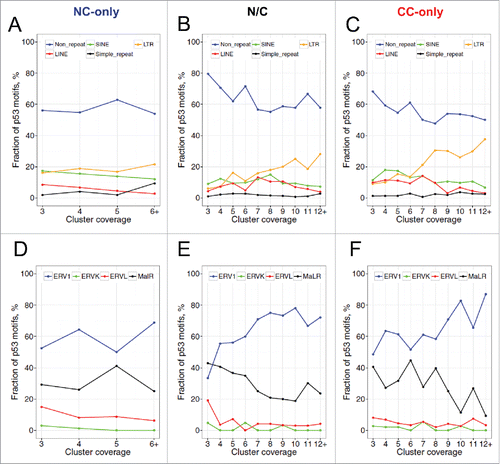
First, the fraction of repeat-associated p53 motifs increases with the coverage of the clusters (). That is, the high-coverage p53 ChIP clusters tend to occur in repetitive regions. Because the high-coverage clusters are likely to have stronger p53 motifs (), it follows that strong p53 motifs are likely to reside in repeats, consistent with earlier studies.Citation45 Second, the fraction of LTR-associated p53 motifs increases with the coverage of the clusters (orange lines in ). Visual inspection of a LTR element on Chromosome 8 showed that a large p53 ChIP cluster (including 14 CC ChIP fragments and 7 NC ChIP fragments) overlap with it (Fig. S9), indicating that this LTR element is a ‘strong’ binding site of p53. Third, the fraction of ERV1-associated p53 motifs increases with the coverage of the clusters (blue lines in ). Overall, about 12% of identified p53 motifs occur in ERV1 elements, which is much higher than the fraction of these elements (2.9%) in the human genome.Citation46 The percentage distribution in repeat families () and subfamilies () is significantly different from the corresponding fractions in the human genomeCitation46 for all groups and all coverage of clusters (Chi-squared test, p < 0.05), except for the pile-3 clusters in .
Overall, our data are generally consistent with the conclusion of previous studies,Citation6 that p53 binding sites are highly enriched in ERV1 repeats. Moreover, we showed that ERV1 is the only repeat family in which NC and CC ChIP clusters are enriched, suggesting that other families such as SINE, LINE and simple repeats may play a less important role in p53 transcriptional programs.
High nucleosome occupancy and DNase I sensitivity at high-confidence p53 binding sites
To examine the chromatin organization around p53 binding sites, we started with 2 well-studied p53 target sites, the 5′ RE and 3′ RE in the CDKN1A/p21 promoter. It has been shown that both p21 REs are located within nucleosomal DNA in vitro and in vivo.Citation26 In such a case, we would expect to see that both 5′ and 3′ REs occur in genomic regions with relatively high nucleosome occupancy in the chromatin landscape of normal (GM12878) and cancer (K562) cell lines. Indeed, we observed increased nucleosome occupancy around the 2 p21 REs (Fig. S10), which is about 2 times higher than the average occupancy in the human genome.
To check whether this tendency holds for the NC and CC p53 ChIP clusters, we calculated in vivo nucleosome occupancy profiles around the ChIP clusters in the NC-only, N/C and CC-only groups. Clearly, for all 3 groups, the centers of ChIP clusters are located in the genomic regions with a significantly higher nucleosome occupancy compared with flanking DNA 1000 bp away (, Wilcoxon test, p < 0.01). To determine whether the observed high nucleosome occupancy is related to intrinsic histone-DNA interaction, we used published data on the positioning of reconstituted human nucleosomesCitation47 and constructed in vitro nucleosome occupancy profiles for the p53 ChIP clusters. The nucleosome occupancy at the cluster centers is much higher compared with flanking DNA (Fig. S11) and this difference is statistically significant (Wilcoxon test, p < 10−15, Table S6). Thus, our results are consistent with the earlier findings that p53 binding sites usually reside within genomic regions with DNA sequences predicted to encode relatively high intrinsic nucleosome occupancy.Citation27
Figure 4. Profiles of nucleosome occupancy (A–C) and DNase I sensitivity (D–F) around p53 ChIP clusters of the NC group (A, D), the N/C group (B, E), and the CC group (C, F). All the ChIP clusters were aligned to their centers (position 0). Nucleosome mapping data and DNase-seq data for GM12878 cells (blue) and K562 cells (red) are shown. The nucleosome occupancy values were normalized with respect to the average value of the genome. The averaged nucleosome occupancy values and DNase-seq values were symmetrized with respect to the centers of the p53 ChIP clusters. The differences between position 0 and positions ± 1000 in each group were evaluated statistically by Wilcoxon tests (Table S6).
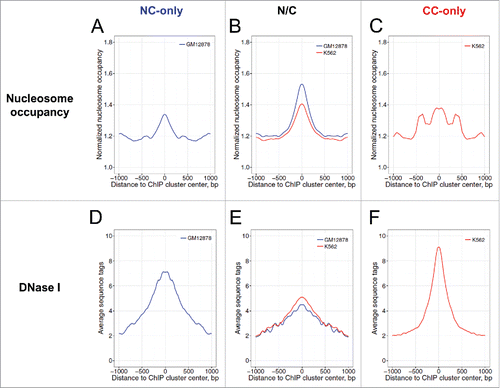
To examine the chromatin accessibility for NC and CC p53 ChIP clusters, we examined the profiles of DNase I sensitivity measured by DNase-seq in GM12878 and K562 chromatin (). We observed that genomic regions near the p53 ChIP clusters exhibit increased DNase I sensitivity compared with flanking DNA (Wilcoxon test, p < 10−15), indicating that, in general, p53 ChIP clusters are located in accessible genomic regions.
In summary, we found that both NC and CC p53 ChIP clusters are characterized by 2 chromatin features. The first is high nucleosome occupancy, which is consistent with earlier reports on p53-induced DNA bending,Citation48,Citation49 and p53 interaction with its target sites exposed on the nucleosomal surface.Citation25,28 This nucleosome-binding ability distinguishes p53 from most TFs, which have functional DNA binding sites that are often found in nucleosome-depleted regions in vivo.Citation21 The second feature is high DNase I sensitivity, which is shared by most TFs.Citation21 Thus, unlike most TFs, p53 binding sites have a unique combination of characteristics, high nucleosome occupancy and high DNase I sensitivity.
Epigenetic landscape around p53 ChIP clusters in normal and cancer cells
To further investigate the chromatin landscape around p53 ChIP clusters, we analyzed the histone modifications and DNA methylation data for GM12878 and K562 cellsCitation50 which we downloaded from the ENCODE database.Citation51 It is well established that lysine methylation of histone H3 and H4 is implicated in either transcriptional activation or repression, depending on the methylation sites. In particular, trimethylation of K4 and K36 on histone H3 (H3K4me3 and H3K36me3) are active histone marks. H3K4me3 is often found at gene promoters, whereas H3K36me3 is associated with transcribed regions in gene bodies.Citation52 By contrast, trimethylation of K27 (H3K27me3) is a repressive signal, primarily found at promoters in gene-rich regions.
We examined the epigenetic profiles of p53 ChIP clusters in the 3 groups and observed several interesting tendencies. First, NC p53 ChIP clusters have a higher level of transcriptionally active histone marks (H3K4me3 and H3K36me3) compared with their counterparts in cancer chromatin ( and Fig. S12). In particular, the NC p53 ChIP clusters in GM12878 chromatin have a significantly higher level of H3K4me3 and H3K36 me3 than the CC ChIP clusters in K562 chromatin (compare and , and Fig. S12A and S12C). For the N/C group, the ChIP clusters in GM12878 chromatin also exhibit a higher level of active histone marks than in K562 chromatin ( and Fig. S12B). Since H3K4me3 and H3K36me3 are often found in active genes (i.e., at promoter and gene body, respectively), our data strongly suggest that p53-DNA interactions in NC chromatin are associated with gene activation. In other words, p53-dependent transcriptional activation is more likely to occur in NC chromatin than in CC chromatin.
Figure 5. Profiles of the active histone mark H3K4me3 (A–C) and the repressive histone mark H3K27me3 (D–F) around the p53 ChIP clusters in the NC-only group (A, D), the N/C group (B, E) and the CC-only group (C, F). The p53 ChIP clusters were aligned to their centers (position 0). The aggregate profiles of H3K4me3 and H3K27me3 data from GM12878 (blue) and K562 (red) are shown for the genomic region around the ChIP cluster centers. The averaged H3K4me3 and H3K27me3 values were symmetrized across the centers. The differences between various groups at position 0 were evaluated statistically by Wilcoxon tests (Table S6).
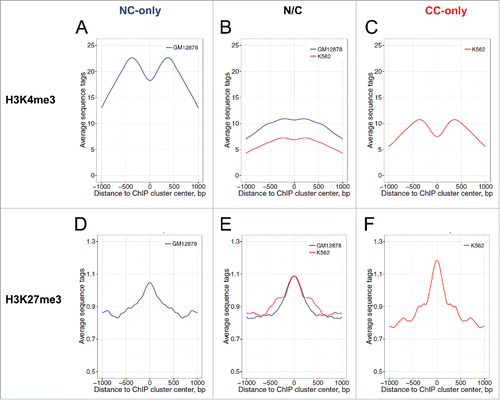
Second, CC p53 ChIP clusters have a significantly higher level of repressive histone mark H3K27me3 than NC ChIP clusters (). These data are consistent with the coupling of p53 binding in CC chromatin with transcriptional repression.
Third, the p53 ChIP clusters in the cancer-only group have a significantly lower level of DNA methylation than their counterparts in the NC-only group ( and ). Localization of the p53 CC -only sites in genomic regions with low DNA methylation may be related to chromatin decondensation due to global DNA hypomethylation in cancer. Unexpectedly, for the p53 ChIP clusters in the N/C group, DNA methylation in K562 chromatin is higher than that in GM12878 chromatin (). Thus, in K562 cancer cell chromatin, the ChIP clusters in the N/C group exhibit a drastically different DNA methylation level from those in the CC-only group ( and , Wilcoxon test, p = 8.0 × 10−13). These data suggest that the interplay between p53 binding and level of DNA methylation in CC chromatin is more complicated than that between p53 binding and histone H3 methylation.
Figure 6. Profiles of DNA methylation around p53 ChIP cluster centers in the NC-only group (A), the N/C group (B) and the CC-only group (C). The MeDIP-seq data of the normal cell line GM12878 (blue) and the cancer cell line K562 (red) were used for this analysis. The MeDIP-seq data at genomic positions flanking the ChIP cluster centers were averaged and symmetrized across the centers (position 0). The differences between various groups at position 0 were evaluated statistically by Wilcoxon tests (Table S6).
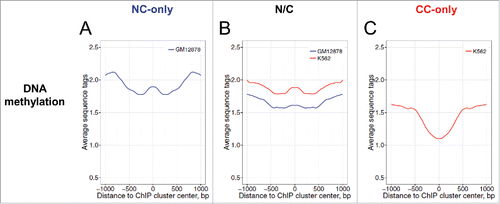
Note that our data differs from the findings of a previous studyCitation18 in 2 aspects. First, Botcheva et al.Citation18 reported that about 40% of p53 binding sites in NC chromatin (IMR90) are located <2 kb from TSSs, while only 15–20% of NC p53 sites were found in the same genomic regions in our study (Fig. S7). Second, Botcheva et al.Citation18 observed that p53 binding sites (in IMR90) are enriched in CpG islands and in hypomethylated DNA, perhaps due to their proximity to TSSs. Although it is highly probable that some NC ChIP clusters occur in hypomethylated DNA, our data indicate that overall, NC ChIP clusters tend to have a high level of DNA methylation (). Note that the Botcheva data (∼800 ChIP fragments) are included in our analysis and account for ∼2% of the whole NC data set (Table S2). The observed differences are probably due to the methods used by Botcheva and colleagues to select the p53 binding sites from their ChIP-seq data.
Discussion
Over-representation of ERV1 family repeats in p53 high-confidence binding sites
As proposed in earlier studies,Citation53 the transposable elements (TEs) are indispensable for evolution of the regulatory networks. `Nowadays, this idea has been supported by the discovery of numerous repetitive elements exapted as cis-regulatory sequences.Citation54 It has been shown that species-specific TEs can greatly shape the transcriptional network of many TFs in humans and mice by contributing up to ∼20% of functional binding sites.Citation55,56
The tumor suppressor p53 is one of the TFs known to utilize TEs to expand its transcriptional networks. We and others have reported the existence of p53 binding sites in primate-specific interspersed repeats, including LTRs,Citation6 SINEs such as Alu,Citation7,Citation8 and LINEs.Citation9 In particular, we have discovered that 15% of the functional p53 REs (24 out of 160) are located in human repeats, and more than half of these repeat-associated REs reside in Alu elements.Citation7 Other studiesCitation6,8 focused on p53 ChIP fragments and found that 30–40% of ChIP fragments reside in repetitive regions. Note that only ∼300 p53 ChIP fragments from a cancer cell line were analyzed in these studies, which only accounts for 0.25% of total fragments analyzed in this work (Table S2). Our analysis showed that about 40% of p53 ChIP clusters occur in various classes of human repeats including SINE, LTR, LINE and simple repeats. We also found that p53 ChIP clusters are enriched in the ERV repeats, in agreement with the conclusion of previous studies.Citation6 Although ERVs only comprise ∼8% of the human genome,Citation46 they account for 30–40% of p53 pile-12+ clusters (). About 70–90% of the LTR-associated p53 motifs reside in ERV1 elements (), highlighting an important role of the ERV1 repeat family in the p53 transcriptional program. Note that the ERV1 family is also over-represented in binding sites of pluripotency factors including OCT4 and NANOG in humans and mice.Citation56 These findings suggest that ERVs act as important transcriptional regulatory elements that may be involved not only in developmental and tissue-specific gene regulation,Citation57 but also in tumor suppression.
It is generally believed that p53 negatively regulates TE transcription and inhibits retrotransposition to maintain genome stability. For example, it was observed that p53, together with DNA methylation, is involved in the epigenetic silencing of TEs.Citation58 In several other studies it was also found that p53 represses the transcription of HERV-1-LTRsCitation59 and Alu elements.Citation60 However, in certain cellular contexts, p53 appears to increase the transcription of LINE-RNAsCitation9 and long intergenic non-coding RNAs (lincRNA) enriched in Alu repeats.Citation61 These studies show that the functional significance of p53 occupancy in the repetitive regions is still far from being clear.
Finally, we found that p53 ChIP clusters with high PWM scores tend to overlap with human repeats ( and ). These data are consistent with Su et al.Citation45 that TE-associated p53 sites have a higher occupancy and correlate with stronger motif sequences, compared with non-TE-associated p53 binding sites. In this study, a strong correlation was described between occupancy and binding strength of p53 motifs.Citation45 In line with this view, our data suggest that the high occupancy of p53 in repetitive regions may be due to the existence of almost perfect p53 motifs in ERV LTRsCitation6 and Alu elements.Citation7
High nucleosome occupancy and DNase I sensitivity: A characteristic chromatin feature of pioneer TFs including p53
The interaction between TFs and their cognate target sites is the central theme of gene regulation. In eukaryotes, DNA wraps around histone octamers to form nucleosomes, which often occlude TF binding. Hence, efficient binding of a given TF to its target site is greatly reduced in the context of chromatin.Citation62 Therefore, a long-held notion is that TFs compete with nucleosomes to gain access to their binding sites.Citation63 This notion was supported by the comprehensive analysis of ChIP fragments bound by 119 human TFs,Citation21 which showed that on average the fragments are located in nucleosome-depleted, DNase I sensitive genomic regions.
However, recent studies have identified a group of TFs known as pioneer factors that can access their target sites in nucleosomal DNA (see ref. Citation23 in which 13 pioneer TFs are listed). Examples of pioneer TFs include FoxA family proteins (FoxA1, FoxA2 and FoxA3) and pluripotency factors (Oct4, Sox2 and Klf4). Earlier studies have suggested that p53-DNA binding also occurs in nucleosome-enriched regions. This idea first came from several observations that (i) p53 induces DNA bending when bound to its targets,Citation48 and (ii) the degree of p53-induced DNA bending correlates with binding affinity – the more pronounced the DNA bend, the higher the affinity of DNA interactions with p5349. Later, both experimental studies,Citation25,Citation26 and computational modelingCitation25 indicated that p53 can interact with its target sites on the surface of a nucleosome. These data are consistent with our observation that p53 binding sites are characterized with high nucleosome occupancy (). Therefore, p53 can be considered as one of the pioneer TFs.Citation23,24
A previous study showed that the 2 well-known p53 REs in the p21 gene reside in the DNase I-resistant domain.Citation29 By contrast, here we show that p53 ChIP clusters as a whole are in DNase I-sensitive regions (). Thus, p53 binding sites exhibit an unusual chromatin pattern: high nucleosome occupancy and high DNase I sensitivity, which distinguishes it from most DNA-binding TFs.Citation21 Note that this unusual chromatin pattern is also observed for the binding sites of the pioneer factors PRCitation30 and FoxA231. Therefore, we suggest that enhanced nucleosome occupancy and DNase I sensitivity may be a general feature of p53 and other pioneer TFs. This unique combination of chromatin features would allow access of pioneer TFs to their binding sites in chromatin before the access of other TFs.
A new model for differential p53 binding to normal and cancer chromatin
We conducted a comprehensive analysis of p53 ChIP clusters from various NC and CC lines. Our work demonstrates that the p53 sites in NC and CC chromatin are characterized by distinct epigenetic features, highlighting the important role of the chromatin context in modulating p53 occupancy in vivo.Citation11,64,65 In light of these findings, we propose a hypothetical model for distinctive p53 binding patterns in NCs and CCs ().
Figure 7. A model for p53 distinctive binding patterns in normal (A) and cancer (B) cells. In normal cells, p53 binding often occurs in genomic regions close to the gene TSS, characterized by high levels of DNA methylation (filled circles) and active histone marks such as H3K4me3 (green dots on histone tails). By contrast, the p53 binding sites in cancer cells are characterized by low levels of DNA methylation (empty circles) and high levels of repressive histone marks (red dots on histone tails). Reduction of DNA methylation levels leads to chromatin decondensation, which helps to expose p53 sites that are normally embedded in closed chromatin domains. Thus, the shift of the genomic binding patterns of p53 in cancer cells appears to reflect cancer-associated epigenetic dysregulation. Both normal and cancer p53 binding sites are enriched in ERV1 retroviral elements.
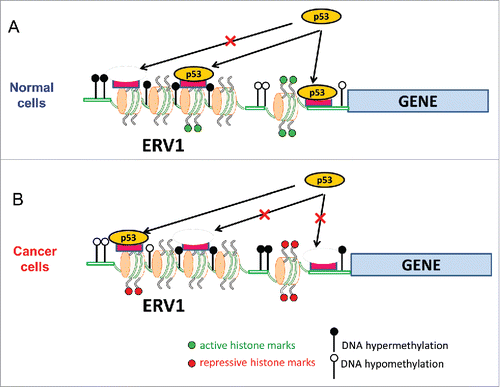
The NC p53 ChIP clusters are often found in genomic regions characterized by high levels of transcriptionally active histone marks such as H3K4me3 and H3K36me3 (). These data are consistent with earlier studiesCitation16 showing that both p53 binding and transactivation are associated with increased H3K4me3 levels. p53-dependent H3K4 methylation is mediated by histone-lysine N-methyltransferase SET1/MLL, which is enhanced by p53- and p300-dependent H3 acetylation.Citation66 Note that H3K4me3 is often found in promoter regions, consistent with our observation that a substantial number (20–25%) of NC p53 binding sites are located <5 kb from a TSS (Fig. S5).
By contrast, the CC p53 ChIP clusters are distinguished by a decreased level of active histone marks (H3K4me3 and H3K36me3) () and an increased level of repressive histone marks (H3K27me3) (Fig. S12). Moreover, the CC p53 sites are characterized by a lower level of DNA methylation, compared with their counterparts in normal cells (), which may be due to genome-wide DNA hypomethylation in CC chromatin.Citation32,33 We hypothesize that global DNA hypomethylation in cancer cells decondenses normally closed chromatin domains, which allows p53 to access new cognate sites in these domains. Taken together, these data suggest that many of these newly exposed p53 binding sites in CC chromatin fall within either genomic regions with repressive histone marks or “proto-enhancers” that are devoid of classic chromatin modifications.Citation24 This may explain why, on average, p53 ChIP clusters in CCs have low levels of active histone marks and high levels of repressive histone marks (). In either case, p53 can engage with its target sites in silent chromatin via pioneer factor activity and, in an appropriate cellular context, can recruit co-factors to activate the enhancers.Citation24 This pioneer activity requires the unique ability of p53 to interact with its target sites in a chromatin state that is non-permissive to most TFs.
Our study provides a simple clue to understanding the distinct p53 genomic binding patterns in normal and cancer cells. That is, p53 binding sites in NCs often occur in active chromatin characterized by H3K4me3 and H3K36me3 marks, which may be coupled with gene activation. By contrast, p53 binding sites in CCs are often found in repressive chromatin characterized by a high level of H3K27me3, which is likely to be associated with gene repression. This model could be tested by the analysis of gene expression data in normal and cancer cells. Understanding p53-dependent gene activation and repression in the context of NC and CC chromatin will shed new light on the complicated cellular mechanisms underlying p53 transcriptional regulation.
Materials and methods
p53 response elements and ChIP fragments
The largest set of functional p53 REs so far has been collected,Citation3 and comprises 156 binding sites. Note, however, that a p53 site from human hepatitis B virus (HBV) and the overlapping sites from the CDKN1 A/p21 gene were included in this data. We removed the HBV site and one of the p21 sites. The final list contains 154 REs. Based on the sequences of the p53 REs, we identified their genomic locations in the hg18 human reference genome (Table S1).
A total of 25 data sets of p53 ChIP fragments were collected (Table S2), including 9 from NC lines and 16 from CC lines. The locations of the fragments were downloaded from the papers and, if needed, ‘converted’ to corresponding locations in the human genome assembly hg18 using the LiftOver utility at the University of California Santa Cruz (UCSC) Genome Browser web server.
Note that the data sets in Table S2 are associated with drug treatments. Only the p53 ChIP fragments caused by certain drug treatment were used for analysis. That is, if a ChIP fragment was found under the untreated (control) condition, it was excluded from analysis even though it was also found under treated conditions. As well, only genomic binding data sets mapped with endogenous wild-type p53 were selected in this study. Those data sets with p53 mutants,Citation67-69 family membersCitation70,71 or p53 in other mammalsCitation17 were not used.
Non-ENCODE nucleosome data sets
Nucleosomes were reconstituted with human granulocyte DNA and recombinantly derived histone octamers to produce a genome-wide in vitro nucleosome map.Citation47 The raw sequence reads from these nucleosomes were downloaded from the NCBI Gene Expression Omnibus (GSE25133). The reads were mapped to human genome assembly hg18 using the Bowtie aligner with the default settings. Only the reads that were uniquely mapped to the genome were used and these reads were extended to 147 bp in the 5′ to 3′ direction. Normalization of nucleosome occupancy across the genome was performed as described before.Citation72 Briefly, for each nucleotide position in the genome, the total number of nucleosomal DNA sequences covering this position was divided by the average number of nucleosomal sequences per base pair across the genome. The resulting value was assigned to this position as the normalized nucleosome occupancy value.
ENCODE MNase-seq, ChIP-seq, MeDIP-seq, and DNase-seq data sets
The MNase-seq reads for in vivo nucleosomes in both GM12878 and K562 cell lines were downloaded from the UCSC Genome Browser FTP server (ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhNsome/). The BAM files in the human genome assembly hg19 were downloaded. The BAM files were then converted to SAM files using SAMtools.Citation73 The reads were then extended to 147 bp in the 5′ to 3′ direction, and the normalized nucleosome occupancy at each position was calculated, as described before.Citation72 The normalized values at each position were smoothed with a 60-bp window.
The ChIP-seq data for histone marks H3K4me3, H3K27me3 and H3K36me3 from both GM12878 and K562 cell lines were downloaded from the UCSC Genome Browser web server (http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19andg=wgEncodeUwHistone). The BAM files in human genome assembly hg19 were downloaded. The BAM files were then converted to SAM files using SAMtools. The start position of each read was shifted by 73 bp in the 5′ to 3′ direction. The values at each position were smoothed twice with a 60-bp window.
DNA methylation MeDIP-seq data and DNase-seq data from both GM12878 and K562 cell lines were downloaded from NCBI GEO data sets (GSM1368906, GSM1368907, GSM816655 and GSM816665). The BigWig files in human genome assembly hg19 were downloaded. The values at each position were smoothed with a 60-bp window.
Since the p53 ChIP clusters are in human genome assembly hg18, the UCSC LiftOver utility was used along with the hg18-hg19 chain file to convert the sites to their corresponding positions in human genome assembly hg19 to make the profiles.
Monte carlo simulation
A Monte Carlo simulation was performed to assess the background level of overlapping ChIP fragments obtained from published studies (Table S2) using BEDTools.Citation74 In the simulation, 42,020 genomic DNA segments (1290 bp on average) and 77,796 segments (663 bp on average) were randomly selected from human genome assembly hg18, and the number of fragments overlapped with others was determined. This process was repeated 100 times to compute the percentage of randomly selected DNA fragments that overlapped. The results are shown in Table S3.
For NC ChIP fragments, we estimated that about 11% of pile-2 clusters, 0.5% of pile-3 clusters and <0.001% of pile-4 clusters can be formed by random sampling. Similar results were obtained for CC ChIP fragments. This suggests that about 88% of the pile-2 and over 99.5% of pile-3 or higher (denoted as pile-3+) clusters likely represent real ChIP enrichment events in normal or cancer cells. Because the pile-2 clusters contain a substantial number of false positive clusters (∼12%), we selected pile-3+ clusters for further analysis.
Repeat analysis
Human repetitive region positions in human genome assembly hg18 were downloaded from the UCSC Genome Browser. The repeat elements were identified using RepeatMasker (v3.2.7) and Repbase Update (9.11). The major types of repeat elements were selected for analysis, including SINE (Alu, MIR), LINE (CR1, L1, L2 and RTE), LTR (ERV1, ERVK, ERVL and Gypsy), Simple Repeat ((TG)n, (TCG)n, (CACTC)n, (GAGTG)n, and (TATATG)n), Low Complexity (C-rich, GC-rich, GA-rich, CT-rich), DNA (MuDR, PiggyBac, TcMar-Mariner, hAT-Charlie). The remaining repeat types were included into the “Other” category.
A p53 motif was classified to be in a repeat (sub)family if it overlaps with any repeat element in that (sub)family. The p53 motifs that are not covered by any repeat elements are classified as a “Non_repeat” group.
Functional annotation
The enriched biologic pathways were identified by DAVID 6.7Citation39,40 using the genes associated with p53 ChIP clusters. If the TSS of a gene was located within 5 kb from the center of a ChIP cluster, this gene was selected for the analysis. Most enriched pathways were determined using DAVID Annotation Chart Analysis and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. All genes associated with the ChIP clusters in the NC-only, N/C and CC-only groups were analyzed by DAVID as a group.
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
KCCY_S_1361064.zip
Download Zip (6 MB)Acknowledgments
The authors are grateful to G. Leiman for editing the text.
Funding
The research was supported by a NIH grant R15GM116102 (to F.C.) and The Intramural Research Program of the National Institutes of Health (Center for Cancer Research, National Cancer Institute) (to V.B.Z.).
Related Research Data
References
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323-331. doi:10.1016/S0092-8674(00)81871-1. PMID:9039259
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat. Genet 1992;1:45-49. doi:10.1038/ng0492-45. PMID:1301998
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402-412. doi:10.1038/nrm2395. PMID:18431400
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497-1502. doi:10.1126/science.1141319. PMID:17540862
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207-219. doi:10.1016/j.cell.2005.10.043. PMID:16413492
- Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, Burgess SM, Brachmann RK, Haussler D. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci USA. 2007;104:18613-18618. doi:10.1073/pnas.0703637104. PMID:18003932
- Cui F, Sirotin MV, Zhurkin VB. Impact of Alu repeats on the evolution of human p53 binding sites. Biol Direct. 2011;6:2. doi:10.1186/1745-6150-6-2. PMID:21208455
- Zemojtel T, Kielbasa SM, Arndt PF, Chung H-R, Vingron M. Methylation and deamination of CpGs generate p53-binding sites on a genomic scale. Trends Genet. 2009;25:63-66. doi:10.1016/j.tig.2008.11.005. PMID:19101055
- Harris CR, Dewan A, Zupnick A, Normart R, Gabriel A, Prives C, Levine AJ, Hoh J. p53 response elements in human retrotransposons. Oncogene. 2009;28:3857-3865. doi:10.1038/onc.2009.246. PMID:19718052
- Beckeman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935. PMID:20679336
- Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell. 2001;8:57-69. doi:10.1016/S1097-2765(01)00283-0. PMID:11511360
- Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243-1254. doi:10.1016/S1097-2765(01)00414-2. PMID:11779500
- Candau R, Scolnick DM, Darpino P, Ying CY, Halazonetis TD, Berger SL. Two tandem and independent subactivation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene. 1997;15:807-816. doi:10.1038/sj.onc.1201244. PMID:9266967
- Vrba L, Junk DJ, Novak P, Futscher BW. p53 induces distinct epigenetic states at its direct target promoters. BMC Genomics. 2008;9:486. doi:10.1186/1471-2164-9-486. PMID:18922183
- Kaneshiro K, Tsutsumi S, Tsuji S, Shirahige K, Aburatani H. An integrated map of p53-binding sites and histone modification in the human ENCODE regions. Genomics. 2007;89:178-188. doi:10.1016/j.ygeno.2006.09.001. PMID:17085012
- Menendez D, Nguyen T, Freudenberg JM, Mathew VJ, Anderson CW, Jothi R, Resnick MA. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013;41:7286-7301. doi:10.1093/nar/gkt504. PMID:23775793
- Li M, He Y, Dubois W, Wu X, Shi J, Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell. 2012;46:30-42. doi:10.1016/j.molcel.2012.01.020. PMID:22387025
- Botcheva K, McCorkle SR, McCombie WR, Dunn JJ, Anderson CW. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle. 2011;10:4237-4249. doi:10.4161/cc.10.24.18383. PMID:22127205
- Botcheva K, McCorkle SR. Cell context dependent p53 genome-wide binding patterns and enrichment at repeats. PLoS One. 2014;9:e113492. doi:10.1371/journal.pone.0113492. PMID:25415302
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008;132:311-322. doi:10.1016/j.cell.2007.12.014. PMID:18243105
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798-1812. doi:10.1101/gr.139105.112. PMID:22955990
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227-2241. doi:10.1101/gad.176826.111. PMID:22056668
- Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014;28:2679-2692. doi:10.1101/gad.253443.114. PMID:25512556
- Sammons MA, Zhu J, Drake AM, Berger SL. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015;25:179-188. doi:10.1101/gr.181883.114. PMID:25391375
- Sahu G, Wang D, Chen CB, Zhurkin VB, Harrington RE, Appella E, Hager GL, Nagaich AK. p53 binding to nucleosomal DNA depends on the rotational positioning of DNA response element. J Biol Chem. 2010;285:1321-1332. doi:10.1074/jbc.M109.081182. PMID:19887449
- Laptenko O, Beckerman R, Freulich E, Prives C. p53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc Natl Acad Sci USA. 2011;108:10385-10390. doi:10.1073/pnas.1105680108. PMID:21606339
- Lidor NE, Field Y, Lubling Y, Widom J, Oren M, Segal E. p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome Res. 2010;20:1361-1368. doi:10.1101/gr.103945.109. PMID:20716666
- Cui F, Zhurkin VB. Rotational positioning of nucleosomes facilitates selective binding of p53 to response elements associated with cell cycle arrest. Nucleic Acids Res. 2014;42:836-847. doi:10.1093/nar/gkt943. PMID:24153113
- Braastad CD, Han Z, Hendrickson EA. Constitutive DNase I hypersensitivity of p53-regulated promoters. J Biol Chem. 2003;278:8261-8268. doi:10.1074/jbc.M204256200. PMID:12475992
- Ballare C, Castellano G, Gaveglia L, Althammer S, Gonzalez-Vallinas J, Eyras E, Le Dily F, Zaurin R, Soronellas D, Vicent GP, et al. Nucleosome-driven transcription factor binding and gene regulation. Mol Cell. 2013;49:67-79. doi:10.1016/j.molcel.2012.10.019. PMID:23177737
- Iwafuchi-Doi M, Donahue G, Kakumanu A, Watts JA, Mahony S, Pugh BF, Lee D, Kaestner KH, Zaret KS. The pioneer transcription factor FoxA maintains an accessible nucleosome configuration at enhancers for tissue-specific gene activation. Mol Cell. 2016;62:79-91. doi:10.1016/j.molcel.2016.03.001. PMID:27058788
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683-692. doi:10.1016/j.cell.2007.01.029. PMID:17320506
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11:726-734. doi:10.1038/nrc3130. PMID:21941284
- Akdemir KC, Jain AK, Alton K, Aronow B, Xu X, Cooney AJ, Li W, Barton MC. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 2014;42:205-223. doi:10.1093/nar/gkt866. PMID:24078252
- McDade SS, Patel D, Moran M, Campbell J, Fenwick K, Kozarewa I, Orr NJ, Lord CJ, Ashworth AA, McCance DJ. Genome-wide characterization reveals complex interplay between TP53 and TP63 in response to genotoxic stress. Nucleic Acids Res. 2014;42:6270-6285. doi:10.1093/nar/gku299. PMID:24823795
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817-825. doi:10.1016/0092-8674(93)90500-P. PMID:8242752
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673-682. doi:10.1016/S1097-2765(01)00213-1. PMID:11463391
- Nozell S, Chen X. p21B, a variant of p21(Waf1/Cip1), is induced by the p53 family. Oncogene. 2002;21:1285-1294. doi:10.1038/sj.onc.1205191. PMID:11850848
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009a;4:44-57
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009b;37:1-13. doi:10.1038/nprot.2008.211. doi:10.1093/nar/gkn923
- Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36:3639-3654. doi:10.1093/nar/gkn232. PMID:18474530
- Gohler T, Reimann M, Cherny D, Walter K, Warnecke G, Kim E, Deppert W. Specific interaction of p53 with target binding sites is determined by DNA conformation and is regulated by the C-terminal domain. J Biol Chem. 2002;277:41192-41203. doi:10.1074/jbc.M202344200. PMID:12171916
- Contente A, Dittmer A, Koch MC, Roth J, Dobbelstein M. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat Genet. 2002;30:315-320. doi:10.1038/ng836. PMID:11919562
- Fischer M, Quaas M, Nickel A, Engeland K. Indirect p53-dependent transcriptional repression of Survivin, CDC25 C and PLK1 genes requires the cyclin-dependent kinase inhibitor p21/CDKN1 A and CDE/CHR promoter sites binding the DREAM complex. Oncotarget. 2015;6:41402-41417. doi:10.18632/oncotarget.6356. PMID:26595675
- Su D, Wang X, Campbell MR, Song L, Safi A, Crawford GE, Bell DA. Interactions of chromatin context, binding site sequence content, and sequence evolution in stress-induced p53 occupancy. PLoS Genet. 2015;11:1004885. doi:10.1371/journal.pgen.1004885
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921. doi:10.1038/35057062. PMID:11237011
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516-520. doi:10.1038/nature10002. PMID:21602827
- Balagurumoorthy P, Sakamoto H, Lewis MS, Zambrano N, Clore GM, Gronenborn AM, Appella E, Harrington RE. Four p53 DNA-binding domain peptides bind natural p53 response elements and bend the DNA. Proc Natl Acad Sci USA. 1995;92:8591-8595. doi:10.1073/pnas.92.19.8591. PMID:7567980
- Nagaich CK, Appella E, Harrington RE. DNA bending is essential for the site-specific recognition of DNA response elements by the DNA binding domain of the tumor suppressor protein p53. J Biol Chem. 1997;272:14842-14849. doi:10.1074/jbc.272.23.14842. PMID:9169453
- Kundaje A, Kyriazopoulou-Panagiotopoulou S, Libbrecht M, Smith CL, Raha D, Winters EE, Johnson SM, Snyder M, Batzoglou S, Sidow A. Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome Res. 2012;22:1735-1747. doi:10.1101/gr.136366.111. PMID:22955985
- ENCODE Project Consortium, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57-74. doi:10.1038/nature11247. PMID:22955616
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343-357. doi:10.1038/nrg3173. PMID:22473383
- Britten RJ, Davidson EH. Gene regulation for higher cells: a theory. Science. 1969;165:349-357. doi:10.1126/science.165.3891.349. PMID:5789433
- Bannert N, Kurth R. Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci USA. 2004;101:14572-14579. doi:10.1073/pnas.0404838101. PMID:15310846
- Bourque G, Leong B, Vega VB, Chen X, Lee YL, Srinivasan KG, Chew JL, Ruan Y, Wei CL, Ng HH, et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008;18:1752-1762. doi:10.1101/gr.080663.108. PMID:18682548
- Kunarso G, Chia NY, Jeyakani J, Hwang C, Liu X, Chan YS, Ng HH, Bourque G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 2010;42:631-634. doi:10.1038/ng.600. PMID:20526341
- Thompson PJ, Macfaclan TS, Lorincz MC. Long terminal repeats: from parasitic elements to building blocks of the transcriptional regulatory repertoire. Mol Cell. 2016;62:766-776. doi:10.1016/j.molcel.2016.03.029. PMID:27259207
- Leonova KL, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, Sen GC, Komarova EA, Gudkov AV. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc Natl Acad Sci USA. 2012;110:E89-E98. doi:10.1073/pnas.1216922110. PMID:23236145
- Chang NT, Yang WK, Huang HC, Yeh KW, Wu CW. The transcriptional activity of HERV-I LTR is negatively regulated by its cis-elements and wild type p53 tumor suppressor protein. J Biomed Sci. 2007;14:211-222. doi:10.1007/s11373-006-9126-2. PMID:17151828
- Stein T, Crighton D, Warnock LJ, Milner J, White RJ. Several regions of p53 are involved in repression of RNA polymerase III transcription. Oncogene. 2002;21:5540-5547. doi:10.1038/sj.onc.1205739. PMID:12165852
- Younger ST, Kenzelmann-Broz D, Jung H, Attardi LD, Rinn JL. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nuclear Acids Res. 2015;43:4447-4462. doi:10.1093/nar/gkv284
- Owen-Hughes T, Workman JL. Experimental analysis of chromatin function in transcription control. Crit Rev Eukaryot Gene Exp. 1994;4:403-441
- Workman JL, Kingston RE. Nucleosome core displacement in vitro via a metastable transcription factor-nucleosome complex. Science. 1992;258:1780-1784. doi:10.1126/science.1465613. PMID:1465613
- Shaked H, Shiff I, Kott-Gutkowski M, Siegfried Z, Haupt Y, Simon I. Chromatin immunoprecipitation-on-chip reveals stress-dependent p53 occupancy in primary normal cells but not in established cell lines. Cancer Res. 2008;68:9671-9677. doi:10.1158/0008-5472.CAN-08-0865. PMID:19047144
- Millau JF, Bandele OJ, Perron J, Bastien N, Bouchard EF, Gaudreau L, Bell DA, Drouin R. Formation of stress-specific p53 binding patterns is influenced by chromatin but not by modulation of p53 binding affinity to response elements. Nucleic Acids Res. 2010;39:3053-3063. doi:10.1093/nar/gkq1209. PMID:21177650
- Tang Z, Chen WY, Shimada M, Nguyen UT, Kim J, Sun XJ, Sengoku T, McGinty RK, Fernandez JP, Muir TW, et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell. 2013;154:297-310. doi:10.1016/j.cell.2013.06.027. PMID:23870121
- Do PH, Varanasi L, Fan S, Li C, Kubucka I, Newman V, Chauhan K, Daniels SR, Boccetta M, Garrett MR, et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012;26:830-845. doi:10.1101/gad.181685.111. PMID:22508727
- Schlereth K, Heyl C, Krampitz AM, Mernberger M, Finkernagel F, Scharfe M, Jarek M, Leich E, Rosenwald A, Stiewe T. Characterization of the p53 cistrome – DNA binding cooperativity dissects p53s tumor suppressor functions. PLoS Genet. 2013;9:e1003726. doi:10.1371/journal.pgen.1003726. PMID:23966881
- Wang B, Niu D, Lam TH, Xiao Z, Ren EC. Mapping the p53 transcriptome universe using p53 natural polymorphs. Cell Death Diff. 2014;21:521-532. doi:10.1038/cdd.2013.132
- Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, Struhl K. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell. 2006;24:593-602. doi:10.1016/j.molcel.2006.10.018. PMID:17188034
- Yang A, Zhu Z, Kettenbach A, Kapranov P, McKeon F, Gingeras TR, Struhl K. Genome-wide mapping indicates that p73 and p63 co-occupy target sites and have similar DNA-binding profiles in vivo. PLoS One. 2010;5:e11572. doi:10.1371/journal.pone.0011572. PMID:20644729
- Cui F, Cole HA, Clark DJ, Zhurkin VB. Transcriptional activation of yeast genes disrupts intragenic nucleosome phasing. Nucleic Acids Res 2012;40:10753-10764. doi:10.1093/nar/gks870. PMID:23012262
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078-2079. doi:10.1093/bioinformatics/btp352. PMID:19505943
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841-842. doi:10.1093/bioinformatics/btq033. PMID:20110278