
ABSTRACT
Cell replication is a fundamental attribute of growth and repair in multicellular organisms. Pancreatic beta-cells in adults rarely enter cell cycle, hindering the capacity for regeneration in diabetes. Efforts to drive beta-cells into cell cycle have so far largely focused on regulatory molecules such as cyclins and cyclin-dependent kinases (CDKs). Investigations in cancer biology have uncovered that adaptive changes in metabolism, the mitochondrial network, and cellular Ca2+ are critical for permitting cells to progress through the cell cycle. Here, we investigated these parameters in the replication-competent beta-cell line INS 832/13. Cell cycle synchronization of this line permitted evaluation of cell metabolism, mitochondrial network, and cellular Ca2+ compartmentalization at key cell cycle stages. The mitochondrial network is interconnected and filamentous at G1/S but fragments during the S and G2/M phases, presumably to permit sorting to daughter cells. Pyruvate anaplerosis peaks at G1/S, consistent with generation of biomass for daughter cells, whereas mitochondrial Ca2+ and respiration increase during S and G2/M, consistent with increased energy requirements for DNA and lipid synthesis. This synchronization approach may be of value to investigators performing live cell imaging of Ca2+ or mitochondrial dynamics commonly undertaken in INS cell lines because without synchrony widely disparate data from cell to cell would be expected depending on position within cell cycle. Our findings also offer insight into why replicating beta-cells are relatively nonfunctional secreting insulin in response to glucose. They also provide guidance on metabolic requirements of beta-cells for the transition through the cell cycle that may complement the efforts currently restricted to manipulating cell cycle to drive beta-cells through cell cycle.
Introduction
Glucose homeostasis in humans is regulated in large part by glucose dependent insulin secretion from pancreatic beta-cells. Insufficient beta-cell number and beta-cell dysfunction underlie both type 1 and 2 diabetes.Citation1-4 While diabetes maybe reversed by beta-cell replacement accomplished by pancreas transplantation, this strategy is impractical for most patients due to a shortage of donors and the requirement for immunosuppression.
Alternatively, there has been interest in fostering endogenous beta-cell regeneration to reverse diabetes. Beta-cell mass increases in infancy through replication but, as beta-cells become more functionally active, their capacity to replicate diminishes, particularly in humans.Citation5 Therefore, there is interest in the regulation of beta-cell cycle with a view to restore beta-cell replication as a means to promote beta-cell regeneration in diabetes. Seminal works and extensive reviews on beta-cell proliferation suggest that the likely missing link in efforts to promote human beta-cell replication is not the failure to express key cell cycle molecules but the failure to activate them.Citation6-10 Most of the studies focused on understanding the requirements for quiescent adult beta-cells to pass the G0/G1 checkpoint and enter the cell cycle,Citation6 have investigated regulatory pathways, such as those dependent on growth factors and cyclin-dependent kinases and their associated cyclins.Citation6,11-13 However, recent works unveiled the importance of pathways regulating the distal cell cycle checkpoints such as the mitotic checkpoint for the survival and proliferation.Citation14 There is also increasing evidence for the integration of cell cycle and metabolism, recently further illustrated by the requirement of regulators of the cell mitotic checkpoint as insulin signaling effectors in hepatic metabolism.Citation15 Moreover, findings available from other mammalian cell types, as well as yeast, have emphasized that successful transition through the cell cycle requires the integration of phases of cell cycle with adaptive changes in both metabolism and the mitochondrial network.Citation16-18
Glucose mediated insulin secretion requires tight linkage between glucose metabolism and mitochondrial oxidative metabolism, with insulin secretion finally driven by transients in cytoplasmic Ca2+.Citation19 These properties of beta-cells are well established in adult humans in which the majority of beta-cells are locked in the G0 phase.Citation20 We hypothesized that, in contrast to the tight constraints on metabolism, mitochondrial function and Ca2+ transients characteristic of beta-cells in G0, cycling beta-cells would exhibit flexibility and adaptability in these same parameters so that they can successfully negotiate cell cycle checkpoints. To test this hypothesis, we employed a well-characterized rat insulinoma cell line (INS 832/13) that is, by definition, replication competent.
In contrast to most prior investigations employing beta-cell lines, we first synchronized the INS 832/13 cells so that the adaptive changes in cell metabolism, mitochondrial network and Ca2+ compartmentalization could be evaluated in relation to the stage of the cell cycle. We established that, in contrast to quiescent beta-cells, replicating INS 832/13 cells do indeed exhibit flexibility and periodicity in metabolism, mitochondrial network structure and Ca2+.
The present studies underscore the importance of taking into account the major changes in metabolism, mitochondrial network and cellular compartmental Ca2+ transients that occur in cell cycle, these parameters being of particular interest in the context of beta-cell biology. Cell lines such as the INS 823/13 cells are frequently used in studies of beta-cell biology, particularly when the techniques require maintaining a monolayer in culture and as such are often used in studies of mitochondrial network and function and cellular Ca2+ transients. Likewise, such cell models have been used to investigate regulation of beta-cell cycle because, unlike primary beta-cells, the cell lines are replication competent. Here, we provide insights into the potential utility of synchronizing beta-cell lines in cell cycle when investigators focus on metabolism, Ca2+ transients and mitochondrial network form and function. Our study alike to the work arising from cancer biology stresses that the interconnectivity and cross-dependence of cell metabolism and cell cycle also extend to beta-cells, and offer further insights as to why replicating beta-cells such as in infancy have limited beta-cell function given the need to coopt metabolism for cell cycle.
Results
Synchronization of INS 832/13 cells at different stages of the cell cycle
Rodent insulinoma cell lines, including INS 832/13, have been used to examine beta-cell replication thanks to their ability to transit through the cell cycle, thus providing possible targets to stimulate proliferation in adult human beta-cells.Citation21 Several studies conducted in yeast and mammalian cells have shown that the cell cycle is synchronized with cellular metabolism. This is to be expected if different stages of the cycle have the specific metabolic requirements needed for the assembly of cellular building blocks into newly formed cells.Citation17,22,23 In most previous studies utilizing insulinoma-derived beta-cell lines to investigate metabolism, mitochondrial morphology, cell function and cellular Ca2+ transients, the cells were unsynchronized.Citation24-28 Because the cells were asynchronous, the averaging of parameters during different stages of the cell cycle most likely concealed transiently induced changes at specific, critical points of the cycle. To overcome this we synchronized INS 832/13 cells at the G1/S, S and G2/M stages of the cell cycle using a synchronization protocol previously used in HeLa cellsCitation23 as depicted in and described in detail in the Materials and Methods.
Figure 1. Synchronization of INS 832/13 cells at different cell cycle stages. (A) INS 832/13 were serum starved (media supplemented with 0.1% fetal calf serum (FCS)) for 56 h, released into cell cycle (media suplemented with 10% FCS), 12 h later treated with aphidicolin for 12 h, then placed in media supplemented with 10% FCS without aphidicolin. Cells were collected at 0 h, 4 h and 12 h after aphidicolin release for flow cytometry analysis. (B) Representative flow cytometry analysis of DNA content in asynchronous and synchronized INS 832/13 at 0 h, 4 h and 12 h post aphidicolin treatment. (C) Representative cell cycle phase distribution of asynchronous and synchronized INS 832/13 collected at 0 h, 4 h and 12 h after aphidicolin release.
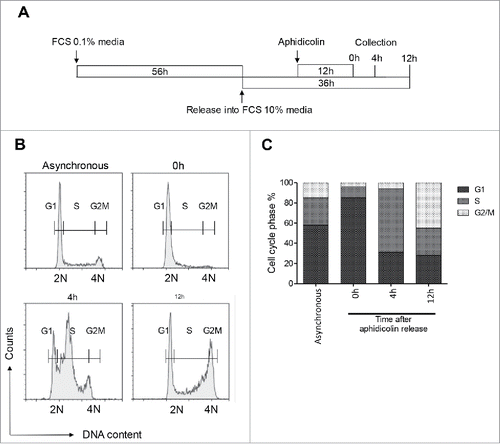
To validate the synchronization protocol in this clonal beta-cell model, we assessed the enrichment of cells in various phases of the cycle through the pattern of known cell cycle regulators by western blot and DNA content by flow cytometry. Representative profiles of DNA content in cells collected at 0, 4 and 12 h post aphidicolin block are presented in and quantified in . Flow cytometry analysis demonstrated that the majority of INS 832/13 cells collected at the 0 h time point were at the G1/S transition, as confirmed by the formation of the early-S phase arm (subG1:0%, G1:58%, S:27%, G2/M:15%; and ). More than half of all cells that were released from aphidicolin block reached the S phase by 4 h post release (63%), and almost half of the cells entered mitosis in a synchronized fashion by 12 h post release (43%; and ). We concluded from these data that the cell populations collected at 0, 4 and 12 h post aphidicolin block were enriched with cells in the G1/S, S and G2/M stages of the cell cycle, respectively.
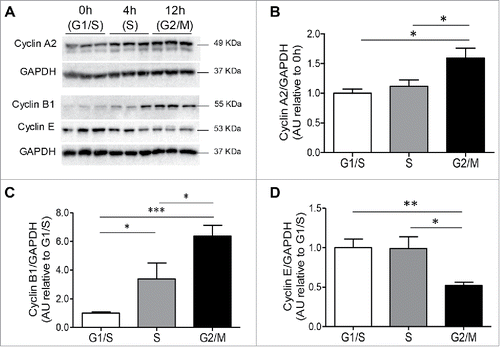
To substantiate the flow cytometry analysis, immunoblotting was used in parallel to measure levels of known molecular markers of the cell cycle. As expected, cyclin A2, a regulator of progression through the cell cycle,Citation29,30 was increased in the S and G2/M phases of the cell cycle (,). Levels of cyclin B1, which is present during the G2/M phase of cell cycle,Citation31 were also most prominent during the G2/M phase of the INS 823/13 cell cycle (p < 0.005 G2/M vs. G1/S; and ). In contrast, cyclin E, a marker of the G1/S transition,Citation16 was increased at G1/S (p < 0.01) and S (p < 0.05) compared to G2/M ( and ). The synchronizing conditions that were used did not induce toxicity, as only a very small (∼1%) subG1 cell population (cells having fragmented DNA indicative of apoptosis) was observed. As expected, all three cyclins were detected in an asynchronous cell population that was studied over a period that was comparable to the synchronized cells (Fig. S1).
Figure 2. Cyclin protein expression in INS 832/13 cells during cell cycle. (A) Protein levels of cyclins were measured by western blot in INS 832/13 cells synchronized at G1/S (0 h post aphidicolin release), S (4 h post aphidicolin release), G2/M (12 h post aphidicolin release). Quantification of Cyclin A2 (B), Cyclin B1 (C) Cyclin E (D) protein levels. Data are presented as mean ± SEM, n = 6 (triplicate from two independent experiments), *p < 0.05, ** p < 0.01, ***p < 0.005.
Having established an effective model of beta-cell synchronization using the INS 832/13 cell line, we next sought to investigate whether changes in beta-cell circadian clock genes, mitochondrial morphology and cell metabolism occurred during the G1/S, S and G2/M stages of the cell cycle.
Transcriptional expression of the circadian clock genes during the cell cycle
There is an increasing focus on the importance of the role of clock genes in regulating cell cycle and in beta-cell function.Citation32-34
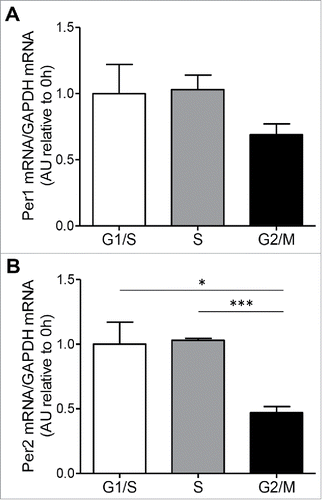
We have evaluated Per1 and Per2 gene expression through the cell cycle ( and ). Interestingly, Per2 mRNA expression levels decreased at the G2/M checkpoint compared to S (p < 0.005) and G1/S phases (p < 0.05; ). Increased Per2 expression has been reported to arrest human leukemia cells at the G2/M checkpoint of cell cycle where they were sensitized to cell deathCitation35 in line with a reported role of Per2 in ATM/ATR associated activation of the G2/M checkpoint in response to damage.Citation36 In the beta-cell model we observed a reduction of Per2 mRNA at the G2/M phase of cell cycle which may suggest a reduction in the circadian-clock-sensitive Wee1 kinase activity thus permitting G2/M transition. Previous studies reported that Per2 loss of function mutations increased the rate of cell proliferation.Citation37,38 Reports indicate that Per2 protects Clock/Bmal1 from cryptochrome (CRY) inhibition Citation39 and consequently increased levels of Clock/Bmal1 arrest mitosis progression through enhanced kinase Wee1 activity.Citation38 The cell cycle-related changes in the expression of the clock gene Per2 mRNA confirmed coupling between cell cycle and circadian clock cycle.
Figure 3. Clock genes transcript levels during cell cycle. (A) mRNA levels of the circadian clock genes, Per1 and Per2 (B), were measured by RT-PCR in INS 832/13 cells synchronized at G1/S (0 h post aphidicolin release), S (4 h post aphidicolin release) and G2/M (12 h post aphidicolin release). Data are presented as mean ± SEM. n = 6 (triplicate from two independent experiments), *p < 0.05, ** p < 0.01, ***p < 0.005.
Mitochondrial morphology and function during the cell cycle
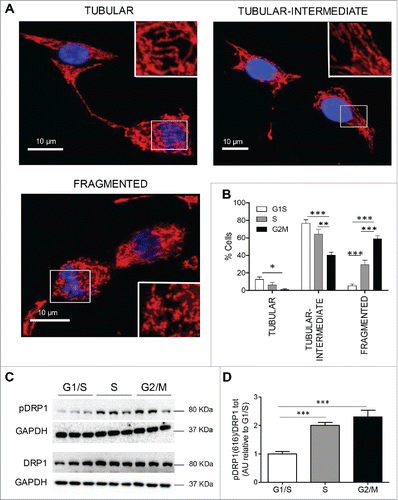
Mitochondria are critical for beta-cell function, integrating the metabolic signals arising from the catabolism of extracellular glucose and transducing these to increased insulin secretion via generation of ATP through oxidative phosphorylation. Mitochondria adapt both functionally and morphologically to the cellular and sub-cellular energetic demands, and exhibit an architecture that ranges from being highly tubular and interconnected, as well as widely distributed throughout the cell, to a more fragmented perinuclear form that is known to be associated with conditions of cell stress.Citation40,41 Consistent with this, the mitochondrial network has been reported as being in a hyperfused state at the G1/S phase of the cell cycle in rat kidney cells and HeLa cells.Citation16,42 In order to test whether the mitochondrial network comparably adapts in beta-cells, we imaged mitochondria in synchronized INS 832/13 cells using Mitotracker Red. Cells were categorized based on the appearance of the mitochondrial network (). At G1/S, almost 80% of the cells had highly tubular mitochondria that became fragmented in the other phases of the cell cycle: 30% of cells in S phase and 60% of cells in G2/M had fragmented mitochondria, respectively (). Asynchronous cells, in contrast, had a variety of mitochondrial forms present (Fig. S2). The shape and form of the mitochondrial network depends on the balance between fusion and fission, processes that are regulated by the dynamin-like GTPases, optic atrophy 1 (Opa1) and dynamin related protein 1 (Drp1). We quantified the expression of these proteins during the cell cycle by Western blot. There was no evidence of altered mitochondrial fusion mediated by Opa1 during the cell cycle as the ratio between long (or active) and short (or inactive) forms of Opa1 did not appear to change (data not shown). However, the fragmentation of the mitochondrial network at the S and G2/M phases of the cell cycle was coincident with the activation of Drp1 by phosphorylation at Ser616 (). The latter increased in the S (p < 0.005 S vs G1/S) and G2/M (p < 0.005 G2/M vs G1/S) stages of the cell cycle () compared to the G1/S.
Figure 4. Changes in mitochondrial morphology at different phases of cell cycle in INS 832/13 cells. (A) 3D projection images of tubular, tubular-intermediate and fragmented mitochondria in synchronized INS 832/13 stained with Mitrotracker Red. Mitochondria were imaged using a confocal Zeiss microscope. (B) Quantification of mitochondrial network in INS 832/13 cells synchronized at G1/S, S and G2/M. Mitochondrial morphology was scored in 250 cells and the data are plotted as mean ± SEM from two independent experiments. (C) Protein levels of DRP1 phosphorylation at Serine 616 and total DRP1 were measured by western blot in INS 832/13 cells synchronized at G1/S, S, G2/M. (D) Quantification of pDRP1(616) and total DRP1 protein levels. Data are presented as mean ± SEM, n = 6 (triplicate from two independent experiments), *p < 0.05, ** p < 0.01, ***p < 0.005.
The activation of Drp1 by phosphorylation at Ser616 depends on the activity of cyclin dependent kinase 1 (Cdk1). The latter in turn requires the regulatory subunit cyclin B1. Cyclin B1 expression during the cell cycle was coincident with increased Drp1 phosphorylation during the S and G2/M phases of the cycle. This is consistent with a previously reported role of cyclin B1 in promoting mitochondrial fragmentation through Drp1 activation in neurons.Citation16,42 These results thus suggest that the cell cycle dependent modification and activation of Drp1 result in mitochondrial fission during DNA synthesis and mitosis.
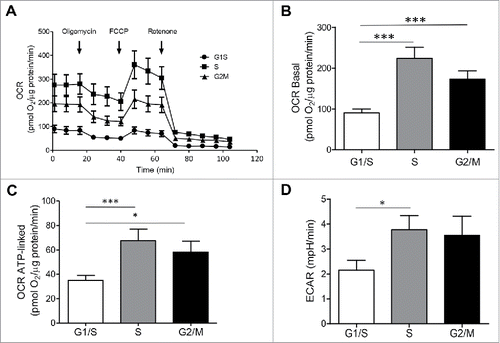
We next asked if the changes in mitochondrial network that we observed during the INS 832/13 cell cycle influence mitochondrial function, potentially facilitating the adaptation of cell metabolism to the requirements of DNA synthesis and mitosis. Mitochondrial membrane potential was relatively unvarying during the progression through different phases of the cell cycle (data not shown), implying that changes in mitochondrial morphology during the cell cycle are an adaptive phenomenon as opposed to a stress response. In contrast to the stable mitochondrial membrane potential we observed, the oxygen consumption rate (OCR) varied through the stages of the cell cycle (). The basal OCR was higher during the S (p < 0.005) and G2/M (p < 0.005) phases in comparison to the G1/S ( and ). A similar pattern was detected in the OCR associated with increased ATP generation (). The basal OCR, the OCR associated with the ATP generation and the ECAR of asynchronous cells were 714 ± 55 pmol O2/μg protein/min, 271 ± 55 pmol O2/μg protein/min and 17 ± 1 mpH/min, respectively. The data are in concordance with previous studies demonstrating that maximal mitochondrial activity occurs during peak DNA synthesis in the S phase.Citation43 Taken together, these findings uncover adaptive changes in the beta-cell mitochondrial network architecture as well as function upon transitioning through the cell cycle, not previously appreciated in cell models utilizing asynchronous beta-cell lines. The fragmentation of the mitochondrial network we observed towards the end of the cell cycle is consistent with the need to separate and sort mitochondria into two daughter cells. The marked increase in OCR in the S and G2/M phases of the cell cycle is consistent with the requirement to synthesize new metabolic intermediates. Given these marked changes in the mitochondrial network and function during the cell cycle, and the known critical role of Ca2+ in beta-cell metabolism, we next evaluated whether the Ca2+ levels of three key subcellular compartments (endoplasmic reticulum, mitochondria and cytoplasm) alter during cell cycle.
Figure 5. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) are increased in INS 832/13 cells synchronized at S and G2/M phases. (A) Oxygen Consumption Rate (OCR) measurements were obtained over time (min) using an extracellular flux analyzer (Seahorse Bioscience). The mitochondrial stress test was used to obtain bioenergetics parameters, by adding the ATP synthase inhibitor Oligomycin A to derive ATP-linked OCR, FCCP to uncouple the mitochondria for maximal OCR, and Rotenone. Bioenergetic profile of INS 832/13 cells in different phases of cell cycle from one experiment (B) Basal oxygen consumption rate (OCR) (C), ATP-linked OCR and (D) ECAR in INS 832/13 cells synchronized at different phases of cell cycle (G1/S, S, G2/M) measured by Seahorse. Data are expressed as mean ± SEM, n = 20 (ten-plicate from two independent experiments), *p < 0.05, ***p < 0.005.
Changes in subcellular Ca2+ distribution during cell cycle
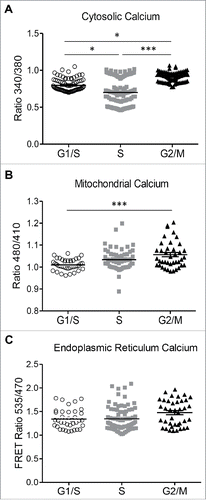
Key steps in the cell cycle are regulated by the interplay of Ca2+ and Ca2+ binding proteins.Citation44 In rat fibroblasts, the levels of Ca2+ in different cell cycle phases vary cyclically.Citation45 Store-operated Ca2+ entry (SOCE), mediated by the interaction between STIM1 (an endoplasmic reticulum Ca2+ sensor) and Orai1 (a cell membrane pore structure), controls the specific checkpoints of cell cycle,Citation46 indicating that stable Ca2+ shifts can regulate cell cycle progression. The fluctuating SOCE activity during cell cycle progression is universal in different cell types, in which SOCE is upregulated in G1/S transition compared to S to G2/M transition. Moreover, Ca2+ transients can also influence cell cycle-specific gene expression.Citation47 We used the fluorescent indicator Fura 2 and Ca2+ sensing probes specifically confined to the ER and mitochondrial matrix to evaluate changes in Ca2+ concentration in different compartments in INS 832/13 cells progressing in a synchronized fashion throughout cell cycle. Adenoviral transduction of Ca2+ probes didn't alter the cell synchrony (Fig S3). The relative Ca2+ levels of the ER were stable during the transition from the G1/S to the G2/M phases of the cell cycle, as monitored by the FRET ratio of an adenoviral ER-targeted probe, D4ER ().Citation48 The levels of cytosolic Ca2+ were highest at G1/S compared to S and G2/M (p < 0.05; ). In contrast, mitochondrial Ca2+ was lowest at G1/S (p < 0.005 vs. S and G2/M; ). This rise in cytosolic Ca2+ at G1/S, apparently through decreased mitochondrial Ca2+ uptake, is consistent with the requirement for Ca2+ to initiate centrosome duplication at early G1/S.Citation49 The rise in mitochondrial Ca2+ during the cell cycle progression in the S and G2/M phases coincided with the increased mitochondrial respiration, presumably by activation of Ca2+-dependent dehydrogenases of the tricarboxylic acid (TCA) cycle. The levels of Ca2+ we observed in ER, mitochondria and cytosol in asynchronous INS 823/13 cells were 1.6 ± 0.04 FRET ratio between 535 nm and 470 nm, 1.1 ± 0.02 ratio between 480 nm and 410 nm and 0.9 ± 0.02 ratio between 340 nm and 380 nm.
Figure 6. Changes in cytosolic and mitochondrial calcium at different phases of cell cycle in INS 832/13 cells. (A) endoplasmic reticulum (ER) (B) cytosolic and (C) mitochondrial calcium concentration was measured in INS 832/13 synchronized at G1/S, S and G2/M by cell imaging using D4ER FRET adenoviral probe, the calcium indicator Fura-2 and ratiometric Pericam respectively. Data are expressed as mean ± SEM, n = 20–30 (ten-plicate from two or three independent experiments), *p < 0.05, ** p < 0.01, ***p < 0.005.
Fluctuations in metabolism during cell cycle
In yeast, the concentrations of critical metabolites exhibit periodic fluctuations during cell cycle, implying that metabolic periodicity may be important for cell cycle progression.Citation18 To assess whether changes in the intracellular metabolites occur in INS 832/13 cells during cell cycle we monitored levels of ∼150 different metabolites in synchronized cells using a mass spectrometry-based approach. Glucose is the major source of energy for beta-cells and regulates insulin secretion. In order to understand the qualitative changes in glucose utilization during the cell cycle, we analyzed the mass isotopologue distribution (MID) of the principal TCA intermediates of INS 823/13 cells using [U-13C6]-labeled glucose as a tracer. Culture media with [U-13C6]-labeled glucose did not alter the synchronization profile of INS 832/13 as shown in Figure S4.
The schematic for the utilization of the glucose tracer by the TCA cycle is depicted in Supplementary Figure S5. In brief, glucose-derived pyruvate (Pyr) has three labeled carbons but loses one C-13 carbon in the conversion to acetyl-CoA, in a reaction catalyzed by pyruvate dehydrogenase (PDH). Then, PDH-derived citrate has two labeled carbons, as do most of the other TCA intermediates, that remain after the end of the first round of the TCA cycle (Fig. S5A). In the next round, citrate now has four labeled carbons and the other downstream intermediates exhibit three or four labeled carbons, depending on the loss of CO2 with a labeled or unlabeled carbon (Fig. S5B). However, Pyr is also converted to oxaloacetate in a reaction catalyzed by pyruvate carboxylase (PC). At this point, malate- and fumarate-derived from oxaloacetate have three labeled and one unlabeled carbon (Fig. S5C).
First, we confirmed that the labeling of glucose metabolized by the cells was efficient, affirmed by the near 100% abundance of its intracellular M6 isotopologue () and, as a consequence, glycolytic metabolites were also labeled close to 100%. Next, we analyzed the labeling pattern of glucose-derived metabolites (M0 – no labeled carbons, M1-M5 – one to five labeled carbons). The M2 fraction of succinate (Suc), fumarate (Fum), malate (Mal) and aspartate (Asp) derived from the conversion of Pyr to acetyl-CoA after the first round of the TCA cycle was increased at G1/S transition (). M3 and M4 fractions of Fum, Mal, Asp were also higher at G1/S compared to the other phases of the cell cycle ( and ). Also in G1/S, the levels of M3 Mal, Asp and Fum were much higher than M3 Suc (), indicating that the M3 fractions of these molecules are derived from the conversion of Pyr to oxaloacetate rather than being produced through the multiple oxidation rounds in the TCA cycle, implying an increased pyruvate anaplerosis. In addition, the presence of M5 citrate (Cit) suggests that oxaloacetate was used at the maximum in consecutive rounds of the TCA cycle during the G1/S stage of the cell cycle ().
Figure 7. Changes of metabolic state of INS 832/13 beta-cells during cell cycle. (A) Glucose mass isotopomer distribution (MID) resulting from culture of synchronized INS 832/13 cells with [U-13C6] glucose for 24 h, (B), M2 fractions of a-ketoglutarate (a-KG), succinate (Suc), fumarate (Fum), malate (Mal), aspartate (Asp), (C), M3 fractions of a-ketoglutarate (a-KG), succinate (Suc), fumarate (Fum), malate (Mal), aspartate (Asp) succinate, (D), M4 fractions of a-ketoglutarate (a-KG), succinate (Suc), malate (Mal), aspartate (Asp), (E), Citrate mass isotopomer distribution (MID) resulting from culture of synchronized INS 832/13 cells with [U-13C6] glucose, (F) M5 fractions of purine nucleoside ADP and nucleotides ATP and GTP, (G), M5 fractions of pirimidine nucleoside CDP and UDP and nucleotides CTP and UTP, (H), Lactate mass isotopomer distribution (MID) resulting from culture synchronized INS 832/13 cells with [U-13C6] glucose. (I) Relative abundance of intracellular metabolites measured in INS 832/13 synchronized at different cell cycle stages (G1/S, S, G2/M) using GC/MS analysis. Data are expressed as mean ± SEM, n = 6 (triplicate from two independent experiments), *p < 0.05, **p < 0.01, ***p < 0.005.
![Figure 7. Changes of metabolic state of INS 832/13 beta-cells during cell cycle. (A) Glucose mass isotopomer distribution (MID) resulting from culture of synchronized INS 832/13 cells with [U-13C6] glucose for 24 h, (B), M2 fractions of a-ketoglutarate (a-KG), succinate (Suc), fumarate (Fum), malate (Mal), aspartate (Asp), (C), M3 fractions of a-ketoglutarate (a-KG), succinate (Suc), fumarate (Fum), malate (Mal), aspartate (Asp) succinate, (D), M4 fractions of a-ketoglutarate (a-KG), succinate (Suc), malate (Mal), aspartate (Asp), (E), Citrate mass isotopomer distribution (MID) resulting from culture of synchronized INS 832/13 cells with [U-13C6] glucose, (F) M5 fractions of purine nucleoside ADP and nucleotides ATP and GTP, (G), M5 fractions of pirimidine nucleoside CDP and UDP and nucleotides CTP and UTP, (H), Lactate mass isotopomer distribution (MID) resulting from culture synchronized INS 832/13 cells with [U-13C6] glucose. (I) Relative abundance of intracellular metabolites measured in INS 832/13 synchronized at different cell cycle stages (G1/S, S, G2/M) using GC/MS analysis. Data are expressed as mean ± SEM, n = 6 (triplicate from two independent experiments), *p < 0.05, **p < 0.01, ***p < 0.005.](/cms/asset/089a8433-66ce-4c72-ad2a-817c0a0406f7/kccy_a_1361069_f0007_b.gif)
Prominent pyruvate anaplerosis at the G1/S stage of the cell cycle would permit an expansion of biomass likely to satisfy the known requirement of cells to reach a sufficient size in order to successfully pass the G1/S checkpoint before DNA replication and mitosis occur.Citation50 The de novo nucleotide synthesis of a new complement of DNA via the pentose phosphate pathwayCitation18 was higher at the S phase, as shown by the enrichment of the M5 isotopologue of purines and pyrimidines nucleosides and nucleotides in this phase ( and ). This glycolysis-dependent synthetic pathway was supported by a high rate of oxygen consumption that is required for the ATP formation needed to sustain nucleotide synthesis ( and ).Citation51 Intracellular lactate levels increased at S and reached a peak at the G2/M phase of the cell cycle (). However, only a small fraction of the lactate produced was generated from the glucose tracer (), as shown by the high levels of the M0 fraction. This indicates that lactate did not originate from the extracellular glucose. Instead, lactate might have been generated from pyruvate that was derived from the malate shuttle.Citation52 and, as such it reflected TCA activity rather than glycolysis. The increased extracellular acidification rate (ECAR) we observed in the S phase implies that protons and lactate were transported out of the cell during this phase of the cell cycle ().
We observed increased levels of palmitate and choline as well, suggesting augmented fatty acid metabolism in the S and G2/M phases of the cell cycle. This increase in lipid metabolism would most likely be indicative of the formation of new phospholipid membranes that would need to be distributed to the two daughter cells.Citation53 () In spite of the increase in mitochondrial respiration associated with ATP generation (Seahorse; ), the relative ATP levels we observed were lower during the S phase (), in agreement with previous reports of enhanced ATP hydrolysis occurring during DNA replication.Citation54 Maintaining a low level of ATP is essential for the unconstrained glycolysis required for DNA synthesis during the S phase,Citation55,56 because high ATP allosterically inhibits the rate–limiting glycolytic enzyme phosphofructokinase 1 (PFK1).Citation56 The increased abundance of oxidized glutathione (GSSG) and α-ketoglutarate (α-KG) in the S and, mainly, the G2/M phases would suggest an increase in oxidative respiration and presumably glutamine anaplerosis of the cell cycle as previously reported in other cell types ().Citation22
In summary, the activities of different metabolic pathways during the INS 832/13 cell cycle were found to be complementary to the requirements of sufficient biomass to pass the G1/S checkpoint, and energy to support DNA and membrane lipid synthesis in S and G2/M. The metabolic changes observed in beta-cells during the cell cycle are listed in and .
Table 1. Changes of metabolic parameters during the cell cycle.
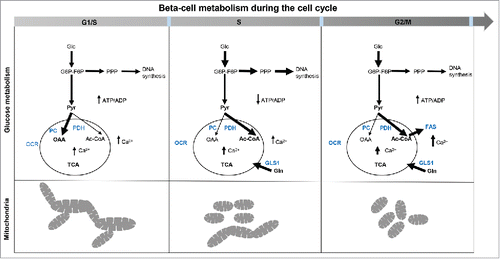
Figure 8. Cellular metabolism in replicating INS 832/13. Schematic of the metabolic changes in INS 832/13 cells synchronized at G1/S, S and G2/M. At G1/S fused-to-intermediate mitochondria dominate and coincide with a peak in the pyruvate anaplerosis; this is alternated in the S phase by a higher involvement of pyruvate dehydrogenase branch of the TCA cycle and more prominent diversion of glucose into the pentose phosphate pathway while at G2/M more fatty acids are synthesized important for securing membrane formation in mitosis and there is another rise in Ca2+ necessary for mitosis. Mitochondria are more fragmented in the S phase and at G2/M phase of the cell cycle to secure mitochondrial inheritance during division.
Discussion
The hypothesis that synchronization of the INS 832/13 beta-cell line in cell cycle would uncover periodicity in metabolism, mitochondrial network and Ca2+ compartmentalization as cells transition through cell cycle was affirmed.
We selected INS 832/13 cells for this study as studies of cell cycle in primary beta-cells are challenging. Aside of the issues of isolating a purified beta-cell preparation, cell replication is infrequent in beta-cells and stability of primary beta-cells in the period of culture required for these studies is also a challenge. Thus, while use of the INS 832/13 cell line rather than primary beta-cells is a limitation of the present studies, fortunately unbiased comparison of metabolic profiles of INS 832/13 cells and primary rat islets indicated that the profiles were quite similar.Citation57,58 These studies suggest that while not perfect, INS 832/13 lines can serve as reasonable model for studies of biochemical features of primary pancreatic beta-cells. The current approaches to induce beta-cell proliferation as a potential therapeutic strategy for diabetes are hampered by the refractoriness of adult beta-cells to replicate, especially in humans. Many previous studies engaged in overcoming this hurdle focused on known regulators of the cell cycle and their effectors.Citation59 Other studies have delineated the human beta-cell G1/S subproteome.Citation6,60 Collectively, these studies affirm that the notoriously replication-deficient phenotype of beta-cells is due either to replication-dependent proteins residing in a non-complementary compartment (i.e. the cytoplasm) and/or the presence of a pro-senescent repertoire of cell cycle inhibitors such as p16 and p27.Citation5
It has long been established in the cancer biology field that proliferating cells have increased energy requirements as they must grow and divide, and that there are specific alterations in the patterns of metabolic provision of energy fuel during cell cycle.Citation22,23 Cell cycle checkpoints not only ensure the genomic integrity of DNA in cells that proceed through the cell cycle, but they also interact with the metabolic pathways of the cell to provide the energy needed for the cell cycle.Citation22,23,61,62 Recently it has been established that the cell cycle checkpoints rely on the metabolic inputs of the energy fuel at the same time being highly synchronized with the circadian rhythm given that the major DNA damage sensors and transmitters such as Chk1 and ATM/ATR are under circadian clock transcriptional regulation.Citation36,63,64 The cell cycle-related changes in the expression of the clock gene Per2 mRNA confirmed coupling between beta-cell cycle and circadian clock cycle. Per2 mRNA expression levels decreased at the G2/M checkpoint. This is logical since Per2 constrains progression through G2/M checkpoint via ATM/ATR activation.Citation36 Furthermore, a reduction in Per2 also facilitates progression in Wee1 kinase dependent manner.Citation64
Since mitochondria play a central role in cellular bioenergetics,Citation16 we examined the mitochondrial network throughout the cell cycle. Beta-cell mitochondria attained a predominantly fused state at the synchronized transition from the G1/S to the S phase and subsequent fragmentation at the G2/M phase of the beta-cell cycle. Mitochondrial fusion at G1/S coincided with a peak in pyruvate anaplerosis, in line with the checkpoint requirements for entry into the S phase and providing energy/currency metabolites (ADP, NADH), repair of damaged mitochondria, complementing mitochondrial DNA, and more importantly preceding maximized ATP production.Citation16,65 Pyruvate anaplerosis is proportional to the conversion of TCA intermediates into the cellular biomass, so that the peak at the G1/S stage of cell cycle is likely synchronized with the requirement of cells to reach a sufficient size and pass the G1/S checkpoint.Citation50 Anaplerosis via pyruvate carboxylase also enables pyruvate cycling, which is required for optimal glucose-mediated insulin secretion by beta-cells, a response that is mediated by ATP generation.Citation66 Therefore pyruvate anaplerosis and mitochondrial fusion at G1/S likely act synergistically to maximize ATP generation prior to the high energy requiring S phase. Interestingly, cytosolic [Ca2+]c was also elevated at the G1/S transition, consistent with the activation of a number of processesCitation67,68 that increase ATP consumption.Citation69,70 This was then matched by an increase in the mitochondrial [Ca2+]m and with this a Ca2+-dependent potentiation of mitochondrial metabolism at G2/M.Citation71
Given that mitochondrial fusion has been demonstrated to contribute to hyperpolarization of the mitochondrial membrane,Citation16 we inquired whether this applies to INS 832/13 cells at G1/S. To our surprise, in contrast to the studies with other cell types, we found that the mitochondrial membrane potential remained constant during all phases of the cell cycle.Citation16,72 Mitochondrial membrane potential is strictly regulated as it is required for the generation of ATP, iron-sulfur cluster biogenesis, import of proteins from cytoplasm and other important functions.Citation73,74 The difference observed in INS 832/13 cells could be inferred by the secretory nature and specific function of beta-cells, where instability of the membrane potential could have adverse effects on beta-cell function. In this sense, the increased mitochondrial activity demonstrated by the higher oxygen consumption rate during the synthesis and division processes in the S and G2/M stages could suggest that more ATP is needed for partitioning beta-cell secretion and beta-cell replication. During the S and G2/M phases of the cell cycle, a high oxygen consumption was complemented by a high flux through glycolysis to support synthesis of a new complement of DNA via the pentose phosphate pathway Citation18 and to generate new lipids required for two daughter cells. Despite the high flux through oxidative phosphorylation during S phase, ATP levels were lowest suggesting a high ATP consumption that physiologically occurs during DNA replication.Citation54
The high mitochondrial activity observed at the S and G2/M stages despite low glucose anaplerosis, suggest utilization of another substrate in the TCA cycle. Colombo et al. (2011) have shown the importance of glutaminolysis in cell progression through S phase of the cell cycle, pointing out that glutamine is critical for providing substrates to the TCA cycle. In our system, the increased levels of α-KG, an intermediate of the glutaminolysis pathway, at S and G2/M would suggest an increased utilization of glutamine by TCA.Citation75 In contrast to other replicating cells, in which glycolysis was shown to be associated with lactate production,Citation75 our MID analysis reveals that only a small fraction of lactate in INS 832/13 cells was derived from the oxidation of glucose with the majority being associated with other cellular processes, such as the malate-pyruvate shuttle.
Taken together, by synchronizing INS 832/13 cells, we have been able to reveal the bioenergetic requirements for the unconstrained progression of beta-cells through the cell cycle. Studies of the functional maturation of beta-cells during development identified a shift of the energy metabolism from a predominantly glycolytic to oxidative phosphorylation,Citation75 revealing the requirement of the metabolic remodeling for the transition from replication propensity to acquisition of functional maturity for glucose-mediated insulin secretion. By implication, it is likely that reversal of these metabolic changes is required to restore the potential for beta-cell replication. Efforts to date to induce replication of adult human beta-cells have focused on the mediators of cell cycle, where as our data imply that such efforts may require simultaneous remodeling of metabolism and synchronization of the cell cycle with the circadian clock. DNA repair programs that sustain cell viability in G0 and G1, may well depend on the permissive or restrictive role of the circadian clock genes so that the cells that traverse the cell cycle in presence of common beta-cell stressors could be sensitized to cell death if any of these complex cell cycle interactions fail due to the injury.
The clinical implications of this are that if beta-cell toxicity occurs under conditions of relatively high beta-cell replication (for example type 1 diabetes in young children), the rate of beta-cell loss will likely be accelerated compared to in adults when beta-cell replication is rare. A related point is that driving quiescent beta-cells into cell cycle under conditions of beta-cell stress (type 1 or 2 diabetes) is likely to result in a decrease rather than increase in numbers of beta-cells.
In conclusion, we have demonstrated that a high level of metabolic-mitochondrial organization during the synchronized beta-cell cycle, supported by Ca2+ transients and adaptive changes in mitochondrial morphology and function. Since beta-cell lines are often used in studies of live cell imaging of Ca2+ and mitochondrial dynamics and as well as studies of beta-cell metabolism, the present studies may be of guidance to future investigators that might wish to synchronize the cells in cell cycle. The latter would reduce the high degree of variance in these parameters between cells when asynchronous in cell cycle. This work highlights the utility of synchronizing cells in the cell cycle when investigating the complex interplay of transition in metabolism, mitochondrial network architecture and function and compartmental Ca2+ transients that are likely required to permit successful cell replication. While INS 832/13 do not represent a perfect model for primary beta-cells,Citation21 the findings represent a first step towards understanding the integration of cell cycle and metabolism in beta-cells.
Experimental procedures
Cell culture
The rat insulinoma cell line INS 832/13 was kindly provided by Dr. C. Newgard (Duke University, Durham, NC, USA). INS 832/13 cells were maintained in RPMI 1640 medium (11.1 mM glucose) supplemented with 10 mM HEPES, 1 mM sodium pyruvate, 100 IU/mL penicillin, and 100 mg/mL streptomycin (Invitrogen, Carlsbad, CA, USA), 10% heat-inactivated fetal bovine serum (Gemini, West Sacramento, CA, USA), and 50 µM β-mercaptoethanol (Sigma, St. Louis, MO, USA) at 37°C in a humidified 5% CO2 atmosphere.
Cell cycle synchronization
Synchronization of cells was carried out according to the scheme presented in . Cells were plated in TCM + 10% FCS and, after 24 h, cells were maintained in TCM + 0.1% FCS for 56 h, to allow them to enter G0. To synchronize cells in G1/S, S and G2/M, medium was changed to TCM + 10% FCS and, 12 h later, aphidicolin, an inhibitor of the B-family DNA polymerases,Citation76 was added. Following 12 h treatment with aphidicolin, the medium was changed to TCM + 10% w/o aphidicolin and cells were collected at 0 h, 4 h and 12 h after aphidicolin release. Cells were determined to be in a specific cell cycle stage based on flow cytometry profiling of DNA content, as mentioned in the Figure legend.
Cell cycle analysis by flow cytometry
Cells were trypsinized, washed in PBS and fixed in 80% methanol at −20°C for at least 2 h. Methanol was removed by centrifugation at 2000 g for 2 min and cells were stained with propidium iodide (50 μg/ml) in the presence of RNase A (50 μg/ml) in PBS for 30 min at 37°C. DNA content analysis was performed using a NovoCyte flow cytometer (ACEA Biosciences, San Diego, CA, USA) equipped with the NovoExpress software.
RNA extraction and qRT-PCR
The levels of Per1 and Per2 mRNA were quantified by qRT-PCR. Total RNA was isolated using RNeasy mini kit (Qiagen) according to the manufacturer's instructions. After denaturation at 65°C, 250 ng of total RNA from each sample was transcribed using SuperscriptIII reverse transcriptase (Invitrogen) at 50°C for 1 h. Real-time quantitative polymerase chain reaction (qPCR) was performed using QuantStudio6 Flex (Applied Biosystems™) with initial denaturation step at 95°C for 20 s, followed by 40 cycles of 94°C for 1 s and 60°C for 20 s. Each qPCR reaction contained 1 × Fast SYBR® Green Master Mix (Applied Biosystems™), 1 μM of each primer, and 400 ng cDNA. The comparative cycle threshold (Ct) method was used to determine the relative mRNA expression of target gene and GAPDH cDNA was used as internal control to normalize the amount of target cDNA. Primer sequences used are as follows: Per1 (rat) F-CCAGGCCCGGAGAACCTTTTT, R-CGAAGTTTGAGCTCCCGAAGT; Per2 (rat) F-CACCCTGAAAAGAAAGTGCGA, R-CAACGCCAAGGAGCTCAAGT; GAPDH (rat) F-ATGACTCTACCCACGGCAAG, R-CTGGAAGATGGTGATGGGTT.
Western blotting
Whole-cell extracts from asynchronous and synchronized cells were prepared using RIPA lysis buffer containing protease inhibitor cocktail (Sigma, St. Louis, MO, USA) followed by sonication for 5 sec. Protein concentration was determined using the DC protein assay kit (Bio-Rad, Irvine, CA, USA). Proteins (30-35 μg/lane) were separated by SDS-PAGE (4–20%) and then transferred onto polyvinylidene fluoride membranes (Bio-Rad, Irvine, CA, USA) by semidry electroblotting. After blocking with 5% milk for 1 h, membranes were probed overnight at 4°C with the following primary antibodies: anti-cyclin A2 polyclonal rabbit (1:1000) (Abcam, ab7956, Cambridge, UK), anti-cyclin B1 polyclonal rabbit (1:1000) (Cell Signaling Technology, 4138 S, Danvers, MA, USA), anti-cyclin E polyclonal rabbit (1:1000) (Santa Cruz, sc-481, Dallas, TX, USA), anti-pDRP1(Ser616) polyclonal rabbit (1:1000) (Cell Signaling Technology, 3455 S, Danvers, MA, USA), anti-DRP1 monoclonal rabbit (1:1000) (Cell Signaling Technology, 8570S, Danvers, MA, USA) and GAPDH monoclonal rabbit (1:1000) (Cell Signaling Technology, 2118S, Danvers, MA, USA). Horseradish peroxidase-conjugated secondary antibodies (1:3000) (Invitrogen, Carlsbad, CA, USA) were applied to the membranes for 1 h at RT. Proteins were visualized by enhanced chemiluminescence (BioRad, Irvine, CA, USA), and protein expression levels were quantified using the Labworks software (UVP).
Mitochondrial network morphology
Cells were grown on glass coverslips in a 6-well plate. Cells were incubated with the cell-permeant mitochondria-specific red fluorescent probe MitoTracker Red CMXRos (Cell Signaling Technology, 9082P, Danvers, MA, USA) at a final concentration of 50 nM in culture medium at 37°C for the last 30 min of culture, washed twice with PBS and fixed in 100% methanol at −20°C for 20 min. Coverslips were then mounted on the glass slides using Vectashield with DAPI, examined under the Axio Imager M2a fluorescence microscope (Zeiss, Oberkochen, Germany) equipped with the imaging system ApoTome.2 and software ZEN2, and imaged using a 63 × objective. For quantitative morphometrical analysis, we obtained images of 250 cells per condition. Mitochondrial morphology was classified as tubular if 100–80% of the cell's mitochondrial area was occupied by tubular, fused mitochondria; tubular-intermediate if 80-40% of the cell's mitochondrial area was occupied by tubular mitochondria; and fragmented if 60–100% of the cell's mitochondrial area was occupied by fragmented mitochondria. Cells in mitosis were presented with extensively fragmented mitochondria. Mitochondrial morphology was independently scored by two different observers (C.M. and K.V.).
Mitochondrial function and glycolysis
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the Seahorse XF Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA). Cells were seeded into the V7 plate (Seahorse Bioscience, North Billerica, MA, USA) at 120,000 cells per well, and the synchronization protocol was followed. To assess the mitochondrial functions, OCR was measured at the basal state and after sequential injection of oligomycin (ATP synthase inhibitor), carbonyl cyanide-p trifluoromethoxyphenylhydrazone (FCCP; uncoupler), and rotenone (complex I inhibitor).
Calcium measurements
Cells were seeded on glass coverslips and synchronized as described above. For cytosolic free calcium measurements, cells were loaded with 2.5 μM fura 2-AM for 45 min in medium containing 11 mM glucose. For mitochondrial and ER measurements, cells were transfected using Lipofectamine 2000 (Mito-Pericam) or infected using adenovirus (D4ER), respectively. For all measurements, cells plated onto coverslips were then transferred to a 1 ml volume perifusion chamber containing 11 mM glucose solution for 7 min, which was followed by 10 minutes of perfusion (0.3 ml/min). Experiments were performed at 32–34° C using inline solution and chamber heaters (Warner Instruments, Hamden, CT). Excitation was provided by a TILL Polychrome V monochromator light source set to 10% of its maximal output. Excitation (x) or emission (m) filters (ETtype; Chroma Technology, Bellows Falls, VT) were used in combination with a FF444/521/608-Di01 dichroic (Semrock Corp., Lake Forest, IL) as follows: Fura-2, 340/10x and 380/10x, 535/30 m (R340x/380x –535 m); D4ER, 430/24x, 470/24 m and 535/30 m (430x – R535 m/470 m); Mito-Pericam, 480/410. Fluorescence emission was collected using a QuantEM:512 SC camera (Photometrics, Tucson, AZ) at 0.125–0.2 Hz. 80–130 cells were analyzed per/sample and data were analyzed using MetaFluor software (Molecular Devices, Sunnyvale, CA).
13C tracing and sample preparation for UHPLC-MS Analysis
For metabolomic analysis, after 56 h in TCM + 0.1% FCS, cells were incubated in TCM + 10% FCS containing [U-13C6] glucose at the concentration of 11.1 mM for 24 h. To extract intracellular metabolites, synchronized cells grown in 6-well plates were briefly rinsed with 2 ml of ice-cold 150 mM ammonium acetate (pH = 7.3). Then, 1 ml of ice-cold 80% MeOH was added per well, and cells were scraped and transferred into Eppendorf tubes. 100 mM D/L-norvaline stock solution was added to reach a 5 nmol final concentration. Samples were vortexed three times for 10 sec maximum, and then spun at 20,000 g for 5 min at 4°C. The supernatant was then transferred into a glass vial, dried using speedvac centrifuge, and reconstituted in 50 μl of 70% acetonitrile (ACN). For mass spectrometry analysis, 5 μl of each sample was injected onto a Luna NH2 (150 mm × 2 mm, Phenomenex, Torrance, CA, USA) column. Samples were analyzed with an UltiMate 3000RSLC (Thermo Scientific, Waltham, MA, USA) coupled to a Q Exactive mass spectrometer (Thermo Scientific, Waltham, MA, USA). The Q Exactive was run with polarity switching (+3.00 kV / −3.00 kV) in full scan mode with an m/z range of 65–950. Separation was achieved using 5 mM NH4 AcO (pH 9.9) and ACN. The gradient started with 15% NH4 AcO and reached 90% over 18 min, followed by an isocratic step for 9 min and reversal to the initial 15% NH4 AcO for 7 min.
Statistical analysis
Results are expressed as the means ± SEM. The statistical analysis was performed by one-way ANOVA using StatView software (SAS Institute Inc., Cary, NC, USA). A value of p < 0.05 was taken as an evidence of statistical significance
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
KCCY_S_1361069.zip
Download Zip (160.8 KB)Funding
This work was supported by funding from the National Institutes of Health (NIH/NIDDK Grant #DK077967) and the Larry Hillblom Foundation (Grant #2014-D-001-NET). Work in the Satin Lab was also supported by #DK46409.
Related Research Data
References
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102-10. doi:10.2337/diabetes.52.1.102. PMID:12502499
- Kloppel G, Drenck CR, Oberholzer M, Heitz PU. Morphometric evidence for a striking B-cell reduction at the clinical onset of type 1 diabetes. Virchows Arch A Pathol Anat Histopathol. 1984;403:441-52. doi:10.1007/BF00737292. PMID:6429944
- Bergman RN, Finegood DT, Kahn SE. The evolution of beta-cell dysfunction and insulin resistance in type 2 diabetes. Eur J Clin Invest. 2002;32(Suppl 3):35-45. doi:10.1046/j.1365-2362.32.s3.5.x. PMID:12028373
- Cerf ME. Beta cell dysfunction and insulin resistance. Front Endocrinol (Lausanne). 2013;4:37. doi:10.3389/fendo.2013.00037. PMID:23542897
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584-94. doi:10.2337/db07-1369. PMID:18334605
- Fiaschi-Taesch NM, Kleinberger JW, Salim FG, Troxell R, Wills R, Tanwir M, Casinelli G, Cox AE, Takane KK, Scott DK, et al. Human pancreatic beta-cell G1/S molecule cell cycle atlas. Diabetes. 2013;62:2450-9. doi:10.2337/db12-0777. PMID:23493570
- Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, Garcia-Ocana A, Stewart AF. Diabetes mellitus–advances and challenges in human beta-cell proliferation. Nat Rev Endocrinol. 2015;11:201-12. doi:10.1038/nrendo.2015.9. PMID:25687999
- Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocana A. Human beta-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes. 2014;63:819-31. doi:10.2337/db13-1146. PMID:24556859
- Stewart AF, Hussain MA, Garcia-Ocana A, Vasavada RC, Bhushan A, Bernal-Mizrachi E, Kulkarni RN. Human beta-cell proliferation and intracellular signaling: Part 3. Diabetes. 2015;64:1872-85. doi:10.2337/db14-1843. PMID:25999530
- Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human beta-cell proliferation and intracellular signaling: Driving in the dark without a road map. Diabetes. 2012;61:2205-13. doi:10.2337/db12-0018. PMID:22751699
- Fatrai S, Elghazi L, Balcazar N, Cras-Meneur C, Krits I, Kiyokawa H, Bernal-Mizrachi E. Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes. 2006;55:318-25. doi:10.2337/diabetes.55.02.06.db05-0757. PMID:16443763
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963-8. doi:10.1172/JCI22098. PMID:15467835
- Tiwari S, Roel C, Wills R, Casinelli G, Tanwir M, Takane KK, Fiaschi-Taesch NM. Early and late G1/S cyclins and Cdks act complementarily to enhance authentic human beta-cell proliferation and expansion. Diabetes. 2015;64:3485-98. doi:10.2337/db14-1885. PMID:26159177
- Shirakawa J, Fernandez M, Takatani T, El Ouaamari A, Jungtrakoon P, Okawa ER, Zhang W, Yi P, Doria A, Kulkarni RN. Insulin signaling regulates the foxM1/PLK1/CENP-A pathway to promote adaptive pancreatic beta cell proliferation. Cell Metab. 2017;25:868-82 e5. doi:10.1016/j.cmet.2017.02.004. PMID:28286049
- Choi E, Zhang X, Xing C, Yu H. Mitotic checkpoint regulators control insulin signaling and metabolic homeostasis. Cell. 2016;166:567-81. doi:10.1016/j.cell.2016.05.074. PMID:27374329
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A. 2009;106:11960-5. doi:10.1073/pnas.0904875106. PMID:19617534
- Tu BP, Kudlicki A, Rowicka M, McKnight SL. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes. Science. 2005;310:1152-8. doi:10.1126/science.1120499. PMID:16254148
- Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, Synovec RE, McKnight SL. Cyclic changes in metabolic state during the life of a yeast cell. Proc Natl Acad Sci U S A. 2007;104:16886-91. doi:10.1073/pnas.0708365104. PMID:17940006
- Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PloS One. 2012;7:e39722. doi:10.1371/journal.pone.0039722. PMID:22829870
- Kushner JA. The role of aging upon beta cell turnover. J Clin Invest. 2013;123:990-5. doi:10.1172/JCI64095. PMID:23454762
- Cozar-Castellano I, Harb G, Selk K, Takane K, Vasavada R, Sicari B, Law B, Zhang P, Scott DK, Fiaschi-Taesch N, et al. Lessons from the first comprehensive molecular characterization of cell cycle control in rodent insulinoma cell lines. Diabetes. 2008;57:3056-68. doi:10.2337/db08-0393. PMID:18650366
- Colombo SL, Palacios-Callender M, Frakich N, Carcamo S, Kovacs I, Tudzarova S, Moncada S. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc Natl Acad Sci U S A. 2011;108:21069-74. doi:10.1073/pnas.1117500108. PMID:22106309
- Tudzarova S, Colombo SL, Stoeber K, Carcamo S, Williams GH, Moncada S. Two ubiquitin ligases, APC/C-Cdh1 and SKP1-CUL1-F (SCF)-beta-TrCP, sequentially regulate glycolysis during the cell cycle. Proc Natl Acad Sci U S A. 2011;108:5278-83. doi:10.1073/pnas.1102247108. PMID:21402913
- Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki JI, Oka Y. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia. 1993;36:1139-45. doi:10.1007/BF00401058. PMID:8270128
- Jhun BS, Lee H, Jin ZG, Yoon Y. Glucose stimulation induces dynamic change of mitochondrial morphology to promote insulin secretion in the insulinoma cell line INS-1E. PloS One. 2013;8:e60810. doi:10.1371/journal.pone.0060810. PMID:23565276
- Nishi Y, Fujimoto S, Sasaki M, Mukai E, Sato H, Sato Y, Tahara Y, Nakamura Y, Inagaki N. Role of mitochondrial phosphate carrier in metabolism-secretion coupling in rat insulinoma cell line INS-1. Biochem J. 2011;435:421-30. doi:10.1042/BJ20101708. PMID:21265734
- Plecita-Hlavata L, Lessard M, Santorova J, Bewersdorf J, Jezek P. Mitochondrial oxidative phosphorylation and energetic status are reflected by morphology of mitochondrial network in INS-1E and HEP-G2 cells viewed by 4Pi microscopy. Biochim Biophys Acta. 2008;1777:834-46. doi:10.1016/j.bbabio.2008.04.002. PMID:18452700
- Spegel P, Sharoyko VV, Goehring I, Danielsson AP, Malmgren S, Nagorny CL, Andersson LE, Koeck T, Sharp GW, Straub SG, et al. Time-resolved metabolomics analysis of beta-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem J. 2013;450:595-605. doi:10.1042/BJ20121349. PMID:23282133
- Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961-71. PMID:1312467
- Yam CH, Fung TK, Poon RY. Cyclin A in cell cycle control and cancer. Cell Mol Life Sci. 2002;59:1317-26. doi:10.1007/s00018-002-8510-y. PMID:12363035
- Pavey S, Russell T, Gabrielli B. G2 phase cell cycle arrest in human skin following UV irradiation. Oncogene. 2001;20:6103-10. doi:10.1038/sj.onc.1204707. PMID:11593418
- Matsuo Y, Campbell PM, Brekken RA, Sung B, Ouellette MM, Fleming JB, Aggarwal BB, Der CJ, Guha S. K-Ras promotes angiogenesis mediated by immortalized human pancreatic epithelial cells through mitogen-activated protein kinase signaling pathways. Mol Cancer Res. 2009;7:799-808. doi:10.1158/1541-7786.MCR-08-0577. PMID:19509115
- Gale JE, Cox HI, Qian J, Block GD, Colwell CS, Matveyenko AV. Disruption of circadian rhythms accelerates development of diabetes through pancreatic beta-cell loss and dysfunction. J Biol Rhythms. 2011;26:423-33. doi:10.1177/0748730411416341. PMID:21921296
- Rakshit K, Qian J, Ernst J, Matveyenko AV. Circadian variation of the pancreatic islet transcriptome. Physiol Genomics. 2016;48:677-87. doi:10.1152/physiolgenomics.00019.2016. PMID:27495157
- Sun CM, Huang SF, Zeng JM, Liu DB, Xiao Q, Tian WJ, Zhu XD, Huang ZG, Feng WL. Per2 inhibits k562 leukemia cell growth in vitro and in vivo through cell cycle arrest and apoptosis induction. Pathol Oncol Res. 2010;16:403-11. doi:10.1007/s12253-009-9227-0. PMID:19957060
- Sancar A, Lindsey-Boltz LA, Kang TH, Reardon JT, Lee JH, Ozturk N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010;584:2618-25. doi:10.1016/j.febslet.2010.03.017. PMID:20227409
- Gu X, Xing L, Shi G, Liu Z, Wang X, Qu Z, Wu X, Dong Z, Gao X, Liu G, et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ. 2012;19:397-405. doi:10.1038/cdd.2011.103. PMID:21818120
- Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255-9. doi:10.1126/science.1086271. PMID:12934012
- Matsumura R, Tsuchiya Y, Tokuda I, Matsuo T, Sato M, Node K, Nishida E, Akashi M. The mammalian circadian clock protein period counteracts cryptochrome in phosphorylation dynamics of circadian locomotor output cycles kaput (CLOCK). J Biol Chem. 2014;289:32064-72. doi:10.1074/jbc.M114.578278. PMID:25271155
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062-5. doi:10.1126/science.1219855. PMID:22936770
- Yu T, Wang L, Yoon Y. Morphological control of mitochondrial bioenergetics. Front Biosci (Landmark Ed). 2015;20:229-46. doi:10.2741/4306. PMID:25553448
- Lee S, Kim S, Sun X, Lee JH, Cho H. Cell cycle-dependent mitochondrial biogenesis and dynamics in mammalian cells. Biochem Biophys Res Commun. 2007;357:111-7. doi:10.1016/j.bbrc.2007.03.091. PMID:17400185
- Darzynkiewicz Z, Staiano-Coico L, Melamed MR. Increased mitochondrial uptake of rhodamine 123 during lymphocyte stimulation. Proc Natl Acad Sci U S A. 1981;78:2383-7. doi:10.1073/pnas.78.4.2383. PMID:6941298
- Whitaker M, Patel R. Calcium and cell cycle control. Development. 1990;108:525-42. PMID:2167196
- Pande G, Kumar NA, Manogaran PS. Flow cytometric study of changes in the intracellular free calcium during the cell cycle. Cytometry. 1996;24:55-63. doi:10.1002/(SICI)1097-0320(19960501)24:1<55::AID-CYTO7>3.0.CO;2-H. PMID:8723903
- Chen YW, Chen YF, Chen YT, Chiu WT, Shen MR. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci Rep. 2016;6:22142. doi:10.1038/srep22142. PMID:26917047
- Azhar M, Krefft M, Saran S, Weeks G, Nanjundiah V. Calcium levels correlate with cell cycle phase and affect the level of the cyclin B transcript in Dictyostelium discoideum. FEMS Microbiol Lett. 1998;161:193-9. doi:10.1111/j.1574-6968.1998.tb12948.x. PMID:9561748
- Ravier MA, Daro D, Roma LP, Jonas JC, Cheng-Xue R, Schuit FC, Gilon P. Mechanisms of control of the free Ca2+ concentration in the endoplasmic reticulum of mouse pancreatic beta-cells: Interplay with cell metabolism and [Ca2+]c and role of SERCA2b and SERCA3. Diabetes. 2011;60:2533-45. doi:10.2337/db10-1543. PMID:21885870
- Matsumoto Y, Maller JL. Calcium, calmodulin, and CaMKII requirement for initiation of centrosome duplication in Xenopus egg extracts. Science. 2002;295:499-502. doi:10.1126/science.1065693. PMID:11799245
- Qu Z, MacLellan WR, Weiss JN. Dynamics of the cell cycle: Checkpoints, sizers, and timers. Biophys J. 2003;85:3600-11. doi:10.1016/S0006-3495(03)74778-X. PMID:14645053
- Marjanovic S, Wielburski A, Nelson BD. Effect of phorbol myristate acetate and concanavalin A on the glycolytic enzymes of human peripheral lymphocytes. Biochim Biophys Acta. 1988;970:1-6. doi:10.1016/0167-4889(88)90215-7. PMID:3370225
- MacDonald MJ. Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J Biol Chem. 1995;270:20051-8. PMID:7650022
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763-77. doi:10.1038/nrc2222. PMID:17882277
- Frigola J, Remus D, Mehanna A, Diffley JF. ATPase-dependent quality control of DNA replication origin licensing. Nature. 2013;495:339-43. doi:10.1038/nature11920. PMID:23474987
- Scholnick P, Lang D, Racker E. Regulatory mechanisms in carbohydrate metabolism. IX. Stimulation of aerobic glycolysis by energy-linked ion transport and inhibition by dextran sulfate. J Biol Chem. 1973;248:5175. PMID:4268695
- Berg T, Guo Y, Abdelkarim M, Fliegauf M, Lubbert M. Reversal of p15/INK4b hypermethylation in AML1/ETO-positive and -negative myeloid leukemia cell lines. Leuk Res. 2007;31:497-506. doi:10.1016/j.leukres.2006.08.008. PMID:17056112
- Spegel P, Andersson LE, Storm P, Sharoyko V, Gohring I, Rosengren AH, Mulder H. Unique and shared metabolic regulation in clonal beta-cells and primary islets derived from rat revealed by metabolomics analysis. Endocrinology. 2015;156:1995-2005. doi:10.1210/en.2014-1391. PMID:25774549
- Gooding JR, Jensen MV, Newgard CB. Metabolomics applied to the pancreatic islet. Arch Biochem Biophys. 2016;589:120-30. doi:10.1016/j.abb.2015.06.013. PMID:26116790
- Chen H, Zhou L, Wan G, Dou T, Tian J. BMI1 promotes the progression of laryngeal squamous cell carcinoma. Oral Oncol. 2011;47:472-81. doi:10.1016/j.oraloncology.2011.03.016. PMID:21482478
- Kohler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ. Cell cycle control of beta-cell replication in the prenatal and postnatal human pancreas. Am J Physiol Endocrinol Metab. 2011;300:E221-30. doi:10.1152/ajpendo.00496.2010. PMID:20978233
- Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297-308. doi:10.1038/nrm2351. PMID:18285803
- Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell. 2005;9:843-54. doi:10.1016/j.devcel.2005.11.006. PMID:16326395
- Kang TH, Leem SH. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 2014;42:4427-34. doi:10.1093/nar/gku094. PMID:24489120
- Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell. 2006;22:375-82. doi:10.1016/j.molcel.2006.03.038. PMID:16678109
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med. 2001;7:934-40. doi:10.1038/90976. PMID:11479626
- Fransson U, Rosengren AH, Schuit FC, Renstrom E, Mulder H. Anaplerosis via pyruvate carboxylase is required for the fuel-induced rise in the ATP:ADP ratio in rat pancreatic islets. Diabetologia. 2006;49:1578-86. doi:10.1007/s00125-006-0263-y. PMID:16752176
- Gall D, Susa I. Effect of Na/Ca exchange on plateau fraction and [Ca]i in models for bursting in pancreatic beta-cells. Biophys J. 1999;77:45-53. doi:10.1016/S0006-3495(99)76871-2. PMID:10388739
- Gilon P, Arredouani A, Gailly P, Gromada J, Henquin JC. Uptake and release of Ca2+ by the endoplasmic reticulum contribute to the oscillations of the cytosolic Ca2+ concentration triggered by Ca2+ influx in the electrically excitable pancreatic B-cell. J Biol Chem. 1999;274:20197-205. doi:10.1074/jbc.274.29.20197. PMID:10400636
- Ainscow EK, Rutter GA. Glucose-stimulated oscillations in free cytosolic ATP concentration imaged in single islet beta-cells: Evidence for a Ca2+-dependent mechanism. Diabetes. 2002;51(Suppl 1):S162-70. doi:10.2337/diabetes.51.2007.S162. PMID:11815476
- Detimary P, Gilon P, Henquin JC. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: A feedback control mechanism in mouse pancreatic islets. Biochem J. 1998;333(Pt 2):269-74. doi:10.1042/bj3330269. PMID:9657965
- Wiederkehr A, Szanda G, Akhmedov D, Mataki C, Heizmann CW, Schoonjans K, Pozzan T, Spat A, Wollheim CB. Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab. 2011;13:601-11. doi:10.1016/j.cmet.2011.03.015. PMID:21531342
- Sweet S, Singh G. Changes in mitochondrial mass, membrane potential, and cellular adenosine triphosphate content during the cell cycle of human leukemic (HL-60) cells. J Cell Physiol. 1999;180:91-6. doi:10.1002/(SICI)1097-4652(199907)180:1<91::AID-JCP10>3.0.CO;2-6. PMID:10362021
- Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709-17. doi:10.1038/sj.cdd.4401231. PMID:12761579
- Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol Cell. 2016; 61:199-209. doi:10.1016/j.molcel.2015.12.002. PMID:26725009
- Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, Bhushan A. DNA methylation directs functional maturation of pancreatic beta cells. J Clin Invest. 2015;125:2851-60. doi:10.1172/JCI79956. PMID:26098213
- Baranovskiy AG, Babayeva ND, Suwa Y, Gu J, Pavlov YI, Tahirov TH. Structural basis for inhibition of DNA replication by aphidicolin. Nucleic Acids Res. 2014;42:14013-21. doi:10.1093/nar/gku1209. PMID:25429975