
ABSTRACT
Threonine synthase (TS) catalyzes the terminal reaction in the biosynthetic pathway of threonine and requires pyridoxal phosphate as a cofactor. TSs share a common catalytic domain with other fold type II PALP dependent enzymes. TSs are broadly grouped into two classes based on their sequence, quaternary structure, and enzyme regulation. We report the presence of a novel zinc ribbon domain in the N-terminal region preceding the catalytic core in TS. The zinc ribbon domain is present in TSs belonging to both classes. Our sequence analysis reveals that archaeal TSs possess all zinc chelating residues to bind a metal ion that are lacking in the structurally characterized homologs. Phylogenetic analysis suggests that TSs with an N-terminal zinc ribbon likely represents the ancestral state of the enzyme while TSs without a zinc ribbon must have diverged later in specific lineages. The zinc ribbon and its N- and C-terminal extensions are important for enzyme stability, activity and regulation. It is likely that the zinc ribbon domain is involved in higher order oligomerization or mediating interactions with other biomolecules leading to formation of larger metabolic complexes.
Introduction
Threonine synthase (TS; EC 4.2.3.1) catalyzes the pyridoxal phosphate (PLP) dependent conversion of O-phosphohomoserine (OPH) and water into threonine and inorganic phosphate as the final step in the threonine biosynthesis pathway.Citation1,2 PLP is one of the biologically active forms of Vitamin B6 and serves as a cofactor for a large number of enzyme families.Citation3 PLP-dependent enzymes have been broadly classified into seven different fold types based on their structures and into three different families based on their reaction chemistries, of which TS is a member of the fold type II and the β-family.Citation3-5
The fold type II of the PLP-dependent enzymes comprises the tryptophan synthase β family proteins that includes TS, tryptophan synthase β subunit, threonine dehydratase, D-/L-serine dehydratase, 1-aminocyclopropane-1-carboxylate deaminase, cystathionine-β-synthase and cysteine synthase (Pfam family PALP, PF00291).Citation4,6 Many of these enzymes are suggested to have specialized from a common generalist ancestor.Citation7,8 The common catalytic core of these enzymes has an α/β 3-layered sandwich architecture that is referred to as the “Tryptophan synthase β subunit-like PLP-dependent enzymes” fold and H-group in SCOP (SCOP identifier 53685)Citation9 and ECOD, respectively.Citation10 There are two sub-domains in the catalytic region, an N-terminal domain with a 4-stranded β-sheet surrounded by α-helices and a C-terminal domain with a 6-stranded β-sheet with flanking α-helices,Citation3 and the overall fold topology resembles that of a Rossmann fold (Rossmann-like X-group of ECOD).Citation10 The active site cleft, with an invariant lysine residue that binds PLP, is located at the interface of the two sub-domains.Citation3
TS is an interesting enzyme as homologs from different organisms have varying quaternary structures and are regulated differently. Based on global sequence alignment, two distantly related classes of TS have been proposed previously.Citation11 Class I or the plant subfamily is suggested to group TS from higher plants, archaebacteria, cyanobacteria and the eubacterial groups of Aquificaceae, Mycobacteria and Bacillus species and possess an additional N-terminal region preceding the conserved catalytic fold.Citation11-13 Class II or the fungal subfamily groups TS from fungi, Proteobacteria, and coryneform eubacteria, and is suggested to differ from the class I TS by the presence of an extended region at their C-termini.Citation11 TS from several organisms belonging to the two classes have been structurally characterized, and the class I and class II TS are seen to exist as homodimers and monomers, respectively.Citation11-18 Dimerization is assisted by swapping of C-terminal secondary structure elements (SSEs) of the class I TS structure.Citation13-15,17 The extended N-terminal region in the plant TS is implicated in the regulation of enzyme activity as it mediates interactions with the allosteric activator, S-adenosyl methionine (SAM).Citation11,15,19,20 In plants, OPH is the branch point intermediate of the methionine and threonine biosynthetic pathways, whereas the bifurcation occurs at an upstream level, viz., that of homoserine in prokaryotes and fungi.Citation20 SAM is the end product of the methionine catabolism and helps in flux coordination between these two biosynthetic pathways by activating TS, thereby partitioning OPH away from methionine biosynthesis when the intracellular levels of methionine and SAM are high.Citation12,21
As plant TS is the only known enzyme besides cystathionine-β-synthase that is allosterically activated by SAM ,Citation14,22 we were interested in studying the provenance of its N-terminal region that plays an important role in this regulation and enzyme stability.Citation11 Our sequence and structure based evolutionary analysis reveal the presence of a zinc ribbon domain in this region. However, we also note the presence of this N-terminal zinc ribbon domain in many TSs belonging to class II and its absence in several class I TSs. This intrigued us, as class II TS function independent of any regulation by SAM. Moreover, we notice an inconsistency in the available literature about the sequentially and structurally equivalent regions in class I and class II TS. Thus, we present a comparative view of all structurally-characterized TSs followed by a detailed analysis of the N-terminal zinc ribbon.
Results and discussion
Comparative structure analysis of class I and class II TS
All TS structures available at the Protein Data Bank (PDB) (, ) possess the PALP core catalytic domain (PF00291). Structures from Thermus thermophilus,Citation13 Mycobacterium tuberculosis,Citation17 Aquifex aeolicus and Arabidopsis thaliana (aTS)Citation14,15 belong to class I and those from Saccharomyces cerevisiae (yTS),Citation12 Escherichia coli (eTS), Brucella melitensis and Burkholderia thailandensisCitation16 belong to class II based on their sequence similarity with TSs initially used for such classification.Citation11 It is interesting to note that though these two classes differ in their quaternary structure, the swapped SSEs, viz., a loop(extended conformation with some α content)-β-α at the C-terminal, involved in dimerization in class I TS are present in all structures. The final resulting fold of the catalytic domain is also identical in both cases. However, the loop is longer in class II TS and guards the region corresponding to the dimerization interface in class I TS.Citation12 Our analysis of the TS structures and their corresponding full-length sequences reveals that there is no additional extended region at the C-terminal of class II TS (). Thus, the originally devised sequence alignmentCitation11 and the proposed lack of equivalence in the C-terminal regionCitation12 are essentially flawed, as also pointed out by another study.Citation17 The extreme evolutionary divergence among the sequences of the two classes of TSs is the likely reason behind not being able to reliably align the sequences independent of the support provided by the respective structures.
Table 1. Available threonine synthase (TS) structures. The table presents a list of all the structurally characterized TSs available at the PDB (release 3 May 2015), corresponding UniProt identifiers, TS class, zinc ribbon domain boundary*, and the organism name. *In case of Arabidopsis thaliana the residue numbers inside brackets “()” indicate the numbers in the PDB structure, while outside ones correspond to the UniProt sequence.
Figure 1. The domain boundaries in representative threonine synthase (TS) structures. Location of the lysine that binds PLP is marked in blue. Equivalent region in all TS joined by colored arrays. Abbreviations: Mt- Mycobacterium tuberculosis, At- Arabidopsis thaliana, Sc- Saccharomyces cerevisiae, Ec- Escherichia coli, Thr_syn_N- Threonine synthase N terminus (PF14821), ZnR- zinc ribbon, PALP- Pyridoxal-phosphate dependent enzyme catalytic domain (PF00291). In the case of A. thaliana, the residue numbers inside brackets “()” indicate the numbers in the PDB structure, while those outside correspond to the UniProt sequence.
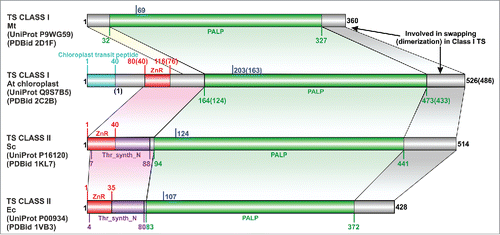
The N-terminal region preceding the PALP catalytic domain in TS is of variable length in different organisms. In class I TS structures from T. thermophilus, M. tuberculosis, A. aeolicus, it is approximately 25 residues long and adopts a predominantly extended conformation. In the structurally characterized class II TS, it is approximately 82 residues and classified as the Pfam “Threonine synthase N terminus” family (Thr_synth_N; PF14821),Citation6 and as the “threonine synthase, domain 1” topology level in CATH (CATH identifier 3.90.1380) under the α-β Complex architecture.Citation23 Current versions of SCOP (v1.75) and ECOD (release 25 January, 2017) databases do not classify this region.Citation9,10 Class I TS from A. thaliana has the longest N-terminal region when compared with other TS structures. Of the 163 residues at its N-terminal, the first 40 residues help in transiting the chloroplast membrane, followed by a characteristic region of about 39 residues that is confined to only TSs from plants as suggested previously.Citation11 This continues into an 84 residue long region that is structurally equivalent to the N-terminal region of class II TS but is not currently classified in Pfam or CATH. As mentioned above (and shown below) this is likely because sequence or even sequence profile-based searches initiated with the sequence of this region from either of the two classes are not able to fetch members of the other class from the pool of non-redundant sequences. Our analysis refutes the previous claims of a completely different fold of the N-terminal domain in both classes,Citation11,12 as besides some conformational changes involving replacement of regular SSEs to extended loop-like structures, the overall fold of the N-terminal region is conserved in all TSs that possess it.
The only unique region in plant TS is the partly disordered region between the chloroplast transit peptide and the common N-terminal region of all TSs, and the SAM binding loop spanning residues 94–106 (PDB identifiers 2C2B, 2C2G).Citation15 The importance of the latter in the allosteric regulation of the enzyme has been emphasized earlier,Citation15 but it is likely that the flexibility exhibited by the plant-specific region is also essential. A superimposition of the two aTS structures reveals that this plant-specific region in the non-SAM bound structures (PDB identifier 2C2G, 1E5X) poses a steric clash with the SAM moieties in the binding pocket in the allosterically activated conformational state (PDB identifier 2C2B). Previous experimental studies that reveal the importance of the residues subsequent to the transit peptide in SAM binding also support this conviction,Citation11,24 and it is plausible that it acts as a lid to regulate the entry of SAM moieties in the binding cavity.
Structural analysis of the common N-terminal region from all TSs
We observe that the common N-terminal region in plant and class II TS structures consists of two segments, viz., a β-rich fragment followed by a α-rich section that connects to the PALP catalytic domain. Though the α-rich part lacks detectable structural similarity with any other known protein folds, we were able to detect striking similarities between the β-rich portion and the zinc ribbon fold (). Zinc ribbons are short protein domains with a structural zinc ion bound by four cysteines contributed by two zinc knuckles.Citation25 Zinc knuckles are short tight turns with a consensus sequence motif CPXCG (X being any aminoacid) and are typically found as turns of β-hairpins. The spacing between the two zinc knuckles is not conserved and can accommodate insertions of variable lengths.Citation25,26 In a large fraction of the zinc ribbon structures, one of the two β-hairpins extends into an additional β-strand that forms a 3-stranded β-sheet with the other β-hairpin.Citation25,27 Depending on the location of the additional β-strand, two circularly permuted forms of the zinc ribbons are known (, ).
Figure 2. Structure and sequence comparison of TS zinc ribbons with bonafide zinc ribbon domains. (A-E) Ribbon diagrams of zinc ribbons from aTS (PDB identifier 2C2B_A), yTS (PDB identifier 1KL7_A), eTS (PDB identifier 1VB3_A), transcription initiation factor IIE α subunit (PDB identifier 1VD4_A), transcription factor IIS (PDB identifier 1TFI_A). In these figures, the N-terminal β-hairpin of the zinc ribbon is colored yellow, the C-terminal β-hairpin is purple, the additional β-strand that forms 3-stranded β-sheet with one of the hairpins is gray, and the zinc knuckles are red. The secondary structure elements (SSEs) that do not constitute the core of the zinc ribbon are colored white. Equivalent SSEs in (A-E) are colored alike. Zinc is shown as an orange sphere, and side chains of zinc-chelating aminoacids and equivalent residues in TS zinc ribbons are represented in stick form. (F) Structure-based multiple sequence alignment of representative TS zinc ribbons and other bonafide zinc ribbons. PDBid/UniProt identifier, organism name abbreviation, the first and the last residue numbers of the regions used in the alignment are indicated for each sequence. Diagrammatic representation of SSEs of the zinc ribbon is indicated above the alignment. Some sequence insertions are not shown, and the numbers of omitted residues are represented by numbers boxed in green. Potential metal-binding ligands are boxed in black and non-metal-binding residues at the same position are boxed in red. Uncharged aminoacids (all except Asp, Glu, Lys, and Arg) in mostly hydrophobic sites are highlighted yellow. Conserved small aminoacids (Pro, Gly) in the vicinity of zinc chelating residues are shown in red. UniProt/PDB identifier and organism name of sequences from eukaryotes, bacteria and archaea are shown in green, black and red, respectively. Sequence stretches where the structures are not superimposable have been italicized. The organism abbreviations are: Sc- Saccharomyces cerevisiae, Ec- Escherichia coli, Bt- Burkholderia thailandensis, Bm- Brucella melitensis, At- Arabidopsis thaliana, Ob- Oryza barthii, Sm- Staphylothermus marinus, Ss- Sulfolobus solfataricus, Ph- Pyrococcus horikoshii, Hm- Haloarcula marismortui, Af- Aliivibrio fischeri, Cc- Campylobacter curvus, Rp- Rhodopseudomonas palustris, Se- Saccharopolyspora erythraea, Gv- Gloeobacter violaceus, Ma- Methanocella arvoryzae, Og- Oceanicola granulosus, Pf- Pyrococcus furiosus, Sy- Synechocystis sp. PCC 6803, Np- Natronomonas pharaonis, Tv- Trichomonas vaginalis, Ca- Chloroflexus aurantiacus, Hp- Helicobacter pylori, Lp- Legionella pneumophila, Pp- Pseudomonas putida, Pa- Pseudomonas aeruginosa.
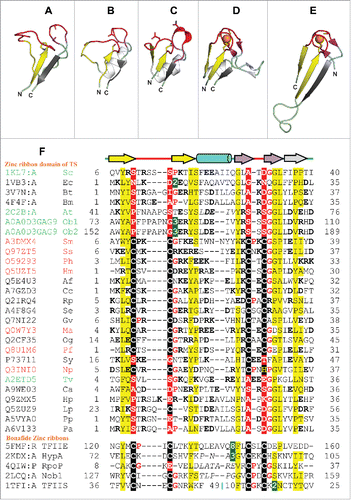
The zinc ribbon domain of TS has a mixed αβ structure with the zinc chelating residues being substituted in all the structurally characterized homologs and thus lacking any bound metal ion. While the eTS () most closely resembles bonafide zinc ribbons (, ) by possessing all the core structural elements and an additional α-helix insertion, in TS structures from all other organisms, most of the core structural elements of the zinc ribbon are replaced by loops having extended conformations. Only the N- and C-terminal β-strands are strictly conserved in all TS zinc ribbons (, , ). The first zinc knuckle in all TS structures is seen having a partly opened up conformation instead of bearing a tight turn. In aTS zinc ribbon, the α-helical insertion seen in class II TS is also substituted by a loop (). None-the-less, the zinc ribbons of aTS and eTS could be manually superimposed with an RMSD of 1.3Å over 29 pairs of Cα atoms.
DALI structure similarity searchesCitation28 initiated with the zinc ribbon domain of any of the TSs recover either themselves or the zinc ribbon domain of other TSs only. This is indicative of the atypical structural characteristics of TS zinc ribbons as compared with bonafide zinc ribbons that make their detection and comparison non-trivial. TopSearch algorithmCitation29 is, however, able to find several zinc ribbon domains in structural similarity searches besides many spurious matches. TopSearch initiated with eTS zinc ribbon (PDB identifier 1VB3_A, residues 1–35) retrieves zinc ribbons from proteins such as transcription initiation factor IIE α subunit (PDB identifier 1VD4_A) with a similarity score of 31.2 and a RMSD of 1.7Å over an alignment length of 33 Cα atoms, and Churchill protein (PDB identifier 2JOX_A) with a similarity score of 31.0, RMSD of 2.2Å over an alignment length of 34 Cα atoms and a single permutation. Similarly, TopSearch initiated with aTS zinc ribbon (PDB identifier 2C2B_A, residues 40–76) is able to retrieve matches to zinc ribbons such as those from E2A DNA-binding protein (PDB identifier 2WAZ_X) with a similarity score of 32.0, RMSD of 2.5Å over an alignment length of 36 Cα atoms and a single permutation, and SIR2 family protein (PDB identifier 5A3B_A) with a similarity score of 30.1 and a RMSD of 2.5Å over an alignment length of 34 Cα atoms. We could manually superimpose the structure of aTS zinc ribbon (PDB identifier 2C2B) and TFIIE (PDB identifier 1VD4) with an RMSD of 1.3 Å over 27 Cα atoms. Likewise, aTS zinc ribbon (PDB identifier 2C2B) and the zinc ribbon of HYPA (PDB identifier 2KDX) could be manually superimposed with an RMSD of 1.5 Å over 23 Cα atoms. The topology of the TS zinc ribbon domain resembles the circularly permuted form seen in sirtuins, rubredoxins and related domains.Citation25
Sequence analysis of the TS zinc ribbon domain
Evidence from sequence analysis corroborates with our structure-based annotation of a zinc ribbon domain in the N-terminal region of TS. A multiple sequence alignment (MSA) of the N-terminal region of TSs in the seed sequences of the Pfam PALP family (PF00291) revealed the presence of cysteine pairs in many organisms. The location of these cysteines corresponds to zinc knuckles of TS and other zinc ribbons (), and these are likely to chelate a metal ion. MSA reveals that many archaea and some bacteria have an intact zinc chelating motif that is lost in homologs from eukaryotes and most bacterial species. The presence of the zinc ribbon domain has also been suggested previously for some archaeal species based on sequence comparisons.Citation30,31 Some homologs also display partial conservation of the zinc chelating residues (e.g. UniProt identifiers A3DKP7, P73711, Q5ZUZ9). Apart from zinc binding residues, conservation of hydrophobic and aromatic residues is seen along the sequence blocks corresponding to β-strands in the structures. A conserved distribution of charged residues is also observed with high propensity at positions +/− 1 or 2 residues surrounding the zinc binding residues, the N- and C-terminal β-strand, and in the region connecting the core β-hairpins of the zinc ribbons.
Sequence comparison using BLAST,Citation32 PSI-BLAST,Citation33 FFAS,Citation34 HMMER and JackHMMER,Citation35 initiated with any of the structurally characterized TS zinc ribbon domain are not able to find statistically significant matches to any other protein domain. However, HMM-HMM based search using HHpredCitation36 initiated with the sequence of aTS zinc ribbon (PDB identifier 2C2B) can retrieve many bonafide zinc ribbons. Matches are found to zinc ribbons from protein such as ribosome biogenesis protein NOP10 (PDB identifier 2APO_B; E-value = 0.00043), transcription initiation factor IIE subunit α (PDB identifier 5FMF_R_B; E-value = 0.0011), hydrogenase/urease nickel incorporation protein HYPA (PDB identifier 2KDX_A; E-value = 0.0039) and DNA-directed RNA polymerase subunit P (PDB identifier 4QIW_A; E-value = 0.003).
Phylogenetic distribution of the TS zinc ribbon
As the zinc ribbon domain is seen to be present in all the available class II TS structures and exclusively in aTS of class I, we set out to study their phylogenomic distribution. We also notice that the zinc ribbon domain is not present in any of the other fold type II PALP protein structures. A phylogenetic tree of type II PALP proteins based on their catalytic domain (Supplementary Figure 1) reveals a separate branch for TSs and related proteins that possess an N-terminal zinc ribbon domain. Further, TSs are seen to partition into two distinct clades, one of which groups TSs from fungi and many Proteobacteria, and corresponds to the conventionally referred class II of TSs.Citation11,37 All of the TSs in this clade possess a zinc ribbon domain, but none have all metal chelating residues preserved. Intriguingly, cysteate synthase that is previously shown to have diverged from TSsCitation31 branches out from this clade and has all four cysteines preserved in the zinc ribbon domain.
The other clade of TS (Supplementary Figure 1) corresponds to the conventional class I and groups plant TS, archaeal TS and many bacterial TS belonging to lineages from Chloroflexi, Planctomycetes, Actinobacteria, Aquificae, Deinococcus-Thermus, Cyanobacteria and some Proteobacteria. This clade displays a selective distribution of TSs that have the PALP domain preceded by a zinc ribbon. For example, Nocardia farcinica (UniProt identifier Q5Z0Z6), Gimesia maris (UniProt identifier A6C359), Oceanicola granulosus (UniProt identifier Q2CF35), all structurally characterized bacterial class I TS with the exception of aTS, and archaeon Thermoplasma volcanium (UniProt identifier Q97CN1) lack a zinc ribbon domain. The conservation of zinc chelating residues also does not follow any pattern, and varying number of substitutions are seen across different branches of the clade. Some other proteins that are not annotated as TS (UniProt identifiers A9B1I1, Q9V0P1, A3DKY8, A2BLL7, A8MBB4) are also seen to branch from this clade, all of which are seen to retain four cysteines in the zinc ribbon domain with the exception of pyridoxal-5-phosphate-dependent protein β subunit (UniProt identifier A9B4P6) that lacks the zinc ribbon.
To specifically study the distribution of TSs possessing a zinc ribbon domain, phylogenetic trees were constructing using the sequences similar to representative sequences from class I and class II, viz., aTS and yTS. As expected, JackHMMER based searches initiated with the zinc ribbon domains of aTS and yTS did not retrieve any common sequences, indicative of an early separation of the two classes. Sequence search initiated with aTS zinc ribbon retrieved sequences from many archaeal lineages (mostly Euryarchaea and some Cren- and Thaumarchaea), bacterial lineages (mostly Chloroflexi, Firmicutes, Planctomycetes, Proteobacteria, Spirochaetes and some Acidobacteria, Actinobacteria, Bacteroidetes, Caldiserica, Cloacimonetes, Cyanobacteria, Elusimicrobia, Fibrobacteres, Gemmatimonadetes, Nitrospinae, Synergistetes, Thermotogae) and eukaryotic lineages (all from Viridiplantae and 2 Rhodophyta- UniProt identifiers M2WSV6, M1VLG3). A phylogenetic tree of full-length representative sequences from this search (Supplementary Figure 2) reveals a single clade of the plant TS, with bacterial homologs as their closest neighbors. Archaeal TS sequences, too, mostly form independent clades, but some (e.g., UniProt identifiers D3SRE7, C7P0 × 4, A0A060HD60) appear to have been laterally acquired from bacteria. Interestingly, we observe the presence of two identical copies of the N-terminal zinc ribbon domain in Oryza barthii (UniProt identifier A0A0D3GAG9). Our sequence similarity searches with aTS zinc ribbon is also able to retrieve some sequences (UniProt identifiers K9WLA2, K8GIS4, Q0EZF2, K7WA11) that are annotated as TS and have a zinc ribbon domain with all four conserved cysteines, but the latter is annotated to belong to the NinF family (PF05810) of the zinc ribbon clan (CL0167) in Pfam.
JackHMMER sequence search initiated with yTS zinc ribbon retrieved mostly bacterial and eukaryotic sequences. The phylogenetic tree of representative sequences (Supplementary Figure 3) shows that class II-like TSs branch into two clades with bacterial and eukaryotic proteins in each clade as suggested previously.Citation38 The only archaeal sequences retrieved were from the Methanomicrobia lineage (UniProt identifiers G7WME5, A0B9I3, K4M8H7, F4BUJ6, A0A0N8WAU2, Q12YN4, L0KU15) and two from the Candidatus Lokiarchaeota lineage (UniProt identifiers A0A0F8VK76, A0A0F8VA59). The bacterial sequences retrieved, mostly belonged to Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria, and some from lineages such as Chlorobi, Cloacimonetes, Cyanobacteria, Deferribacteres, Deinococcus-Thermus, Elusimicrobia, Fusobacteria, Gemmatimonadetes, Ignavibacteriae, Lentisphaerae, Nitrospirae, Planctomycetes, Spirochaetes, Thermodesulfobacteria, and Verrucomicrobia. Eukaryotic matches were mostly confined to the fungal and metazoan lineages, and some from Viridiplantae, Alveolata, Amoebozoa, Cryptophyta, Euglenozoa, Haptophyceae, Heterolobosea, Opisthokonta, Rhizaria, and Stramenopiles. The metazoan sequences retrieved included two TSs from Homo sapiens (UniProt identifiers Q8IYQ7, Q86YJ6). This finding supports a previous report of TS homologs (though with alternate functions) in mammalsCitation38 and thus, conflicting the view of considering TS as a potential drug target.Citation17,39
Discussion
Based on sequence and structural analysis we report a zinc ribbon domain at the N-terminal of the catalytic domain of TS. Co-occurrence of zinc ribbons and domains with a Rossmann-like fold is seen in many proteins such as aminoacyl-tRNA synthetases, adenylate kinases, sirtuins and methyltransferases.Citation25 The zinc ribbons in these proteins, and the linker joining it to the core catalytic domain plays important structural and functional roles.Citation40-43 We observe that like in TSs, the zinc ribbon domain precedes the catalytic domain in several methyltransferases that bind SAM, though the latter acts as a substrate in those proteins (e.g., PDB identifiers 1P91, 3ND1, 4RVD).Citation41,42,44 The zinc ribbon domain in rRNA large subunit methyltransferase (RlmA) is suggested to be important for the recognition and binding specificity of the enzyme toward certain rRNA substrates,Citation42 and the linker joining the zinc ribbon to the core catalytic domain in D-mycarose 3-C-methyltransferase is shown to carry a crucial tyrosine residue that acts as a lid to cover the substrate during catalysis.Citation41
Drawing parallels from the above quoted examples, the zinc ribbon domain of TS is also likely to play an important role in the enzyme stability and function. Indeed, in either case, where it binds zinc or otherwise, the N-terminal zinc ribbon containing region is shown to be essential for enzyme stability.Citation11,31 Its presence has been shown to be crucial for enzyme expression, folding and acquisition of active quaternary structure in TS and the evolutionarily related cysteate synthase in Methanosarcinales.Citation31 Our analysis of the aTS structure (PDB identifier 2C2B) reveals that the catalytic PALP domain has a large hydrophobic patch that is shielded by the zinc ribbon domain (Supplementary Figure 4). Thus, a probable evolutionary reason that justifies the fusion of PALP catalytic domain with a zinc ribbon in TSs is that the latter helps prevent any aggregation that might occur due to hydrophobic interactions.
Further, we predict that the zinc ribbon domain in TSs, like most zinc finger domains, may be involved in mediating homomeric interactions with itself or heteromeric interactions with other biomolecules such as proteins and nucleic acids leading to formation of higher order oligomers or larger metabolic complexes. The crystal packing of the dimeric aTS (PDB identifier 2C2B) reveals close placement of one of the zinc ribbon domain in each dimer to the catalytic domain and the C-terminal region of the other dimer, indicative of a plausible higher level oligomerization that may be achieved as assisted by the zinc ribbon domain. This assumption is supported by a report that shows that truncation of the zinc ribbon from TS and cysteate synthase of Methanosarcina acetivorans results in loss of the functional trimeric form.Citation31 Different oligomeric forms of TSs, viz. monomeric in yTS, dimeric in aTS and trimeric in some archaeal TSs, have been suggested to be related to the differences in their allosteric regulation.Citation31 The removal of the zinc ribbon domain containing region from the aTS has been also shown to result in loss of sensitivity to the allosteric activator SAM and increased susceptibility toward inhibition by substrate analogs.Citation11
We observed conservation of zinc-chelating residues in most archaeal TSs and several bacterial species belonging to class I TSs. Most eukaryotes lack residues for metal chelation in the zinc ribbon domain as observed from their sequences. The N-terminal zinc ribbon containing TS are seen to be widely distributed across all three domains of life and in both classes of TSs. Our analysis suggests that the PALP catalytic domain fused with an N-terminal zinc ribbon was the original ancestral state of TS. Selective loss of the complete zinc ribbon domain seen in specific lineages might have occurred later in evolution. This hypothesis is additionally supported by the fact that the common branching point i.e., OPH in the methionine and threonine biosynthesis pathways seen in plants is regarded as the ancestral state that shifted to the upstream homoserine level later during evolution.Citation45,46 We predict that the zinc ribbon, its N-terminal extension (seen only in extant plant TS) and the linker joining it to the catalytic domain were likely important for the modulation of the enzymatic activity in the ancestral state. The shift in the common branch point has concomitantly affected the sequence and structure of these functionally important regions, including the loss of the plant specific N-terminal ∼40 residues and the SAM-binding loop in the linker region from other organisms. Nonetheless, most extant TSs still possess the zinc ribbon domain that is tightly associated with the catalytic domain and plausibly indispensable for enzyme stability, activity and regulation.
Methods
Structure analysis
Fourteen TS structures were retrieved from the PDB (release 3 May 2016). The structures were visualized and manually compared using the molecular visualization program PyMOL. DaliCitation28 (against PDB release 3 May 2016) and TopSearchCitation29 (against PDB release 27 Apr 2016) tools were used to evaluate the structural similarity of TS zinc ribbon domain with other protein structures. Manual superimpositions of zinc ribbon domains were performed in PyMOL by defining the equivalent regions using the pair fitting command.
Delineating domain architectures
Domain architectures () are drawn using DOG (Domain Graph, version 1.0) software.Citation47 Domain boundaries for the catalytic domain (PALP; PF00291) and threonine synthase N-terminal region (Thr_synth_N; PF14821) were determined by referring to Pfam v. 30.0.Citation6 Zinc ribbon domain boundary was manual delineated, and the chloroplast transit peptide is as defined by UniProt.Citation48
Sequence analysis
The HHpred serverCitation36 (against PDB70_02May16, E-value threshold of 0.001, using MSA generation method HHblits run for 5 iterations) was used to evaluate the sequence similarity of the TS zinc ribbon with other proteins in the PDB. Structure-guided MSA of the TS and other bonafide zinc ribbons was done manually. MSA of TS homologs was made using the ClustalW program (Larkin et al., 2007) within the BioEdit software package (version 7.2.2) (Hall, 1999) with default parameters. Any misaligned regions in the automatically generated MSA were adjusted manually.
Phylogenetic analysis
All the phylogenetic trees were constructed using Maximum Likelihood (ML) method based on the Jones et al. w/freq. modelCitation49 with 100 bootstrap replicates. A discrete Gamma distribution with invariable sites was used to model evolutionary rate differences. All positions containing gaps and missing data were used for tree construction. All phylogenetic analyses were conducted in MEGA6.Citation50 The protein domain architectures and taxa colors were added to the final tree using iTOL.Citation51 The domain boundaries were obtained by referring to the Pfam,Citation6 UniProtCitation48 and Conserved Domain Database (CDD).Citation52 Details of the sequences used and the initial tree construction method for the heuristic search in the individual cases are as below.
The phylogenetic tree of all fold type II PALP dependent enzymes was constructed based on the common catalytic domain in the Pfam “PALP” family (PF00291). A total of 151 sequences were used for the tree construction of which 143 were from the Pfam seed sequences (3 obsolete UniProt entries were discarded from the 146 available at Pfam) and 8 were of the structurally characterized TSs. Pfam seed sequences alignment was considered as the master alignment and the sequences of TS catalytic domain were manually aligned to it guided by the structure superimpositions. Initial tree(s) for the heuristic search were made automatically based on the Neighbor-Join (NJ) and BioNJ algorithms applied to a matrix of pairwise distances estimated using a JTT model, and thereafter accepting the tree topology with a superior log likelihood value.
For constructing the phylogenetic tree of sequences of class I TS with zinc ribbons, sequences were obtained by running 12 iterations of JackHMMER at the HMMER web serverCitation35 using aTS zinc ribbon (PDB identifier 2C2B, residues 40–76) against the Representative Sets (UniProt) rp55 v. 2016–03–02, E-value cut-off: 0.01. Of the 390 sequences obtained in the search, unrelated zinc fingers, and very short/long sequences that cause misalignments were removed, and the remaining 350 were clustered using cd-hitCitation53 at 60% sequence identity and 90% sequence coverage. The resulting 94 sequences were used to construct MSA using the MUSCLE programCitation54 with default parameters from within the MEGA6 software package.Citation50 The initial tree(s) for the heuristic search were obtained automatically by applying the Maximum Parsimony (MP) method.
For constructing the phylogenetic tree of sequences of class II TS with zinc ribbons, sequences were obtained by running 11 iterations (until convergence) of JackHMMER at the HMMER web serverCitation35 using yTS N-terminal zinc ribbon region (PDB identifier 1KL7, residues 1–41) against the Representative Sets (UniProt) rp55 v. 2016–03–02, E-value cut-off: 0.01. Of the 2233 sequences obtained in the search, a single unrelated zinc finger sequence was removed, and the remaining were clustered using cd-hitCitation53 at 40% sequence identity and 90% sequence coverage. From the resulting 123 sequences, short sequences, and protein fragments were removed, and a total of 102 sequences were used to construct MSA using the MUSCLE programCitation54 with default parameters from within the MEGA6 software package.Citation50 The initial tree(s) for the heuristic search were obtained automatically by applying the MP method.
Disclosure of potential conflicts of interest
Authors declare no competing interests.
Authors' contributions
GK did the sequence, structure and phylogenetic analysis of the zinc ribbon domain in threonine synthase and wrote the manuscript. SS supervised the study. Both authors read and approved the manuscript.
Supplemental Files
Download Zip (1.6 MB)Acknowledgments
This work was supported in part by the Department of Biotechnology (project BTISNET; GAP001), XII Five-year plan network project GENESIS (BSC0121) and intramural funds (OLP_0072) from the Council of Scientific and Industrial Research (CSIR) – Institute of Microbial Technology, Chandigarh, India. G.K. is supported by the Shyama Prasad Mukherjee Fellowship of CSIR, India. The authors are grateful to Ms. Indu Khatri for help in the initial phase of this study and Dr. Aravind Iyer of NCBI for his comments on the functional implications of the zinc ribbon in TSs.
Related Research Data
References
- Flavin M, Slaughter C. Purification and properties of threonine synthetase of Neurospora. The Journal of biological chemistry. 1960;235:1103-8. PMID:13823379
- Cohen GN, Hirsch ML. Threonine synthase, a system synthesizing L-threonine from L homoserine. Journal of bacteriology. 1954;67:182-90. PMID:13129211
- Schneider G, Kack H, Lindqvist Y. The manifold of vitamin B6 dependent enzymes. Structure. 2000;8:R1-6. doi:10.1016/S0969-2126(00)00085-X. PMID:10673430.
- Grishin NV, Phillips MA, Goldsmith EJ. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein science: a publication of the Protein Society. 1995;4:1291-304. doi:10.1002/pro.5560040705. PMID:7670372
- Alexander FW, Sandmeier E, Mehta PK, Christen P. Evolutionary relationships among pyridoxal-5′-phosphate-dependent enzymes. Regio-specific alpha, beta and gamma families. European journal of biochemistry / FEBS. 1994;219:953-60. doi:10.1111/j.1432-1033.1994.tb18577.x.
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. The Pfam protein families database: towards a more sustainable future. Nucleic acids research. 2016;44:D279-D85. doi:10.1093/nar/gkv1344. PMID:26673716
- Parsot C. Evolution of biosynthetic pathways: a common ancestor for threonine synthase, threonine dehydratase and D-serine dehydratase. The EMBO journal. 1986;5:3013-9. PMID:3098560
- Parsot C. A common origin for enzymes involved in the terminal step of the threonine and tryptophan biosynthetic pathways. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:5207-10. doi:10.1073/pnas.84.15.5207. PMID:3110785
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. Journal of molecular biology. 1995;247:536-40. doi:10.1016/S0022-2836(05)80134-2. PMID:7723011
- Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, Kim BH, Grishin NV. ECOD: an evolutionary classification of protein domains. PLoS computational biology. 2014;10:e1003926. doi:10.1371/journal.pcbi.1003926. PMID:25474468
- Laber B, Maurer W, Hanke C, Grafe S, Ehlert S, Messerschmidt A, Clausen T. Characterization of recombinant Arabidopsis thaliana threonine synthase. European journal of biochemistry / FEBS. 1999;263:212-21. doi:10.1046/j.1432-1327.1999.00487.x
- Garrido-Franco M, Ehlert S, Messerschmidt A, Marinkovic S, Huber R, Laber B, Bourenkov GP, Clausen T. Structure and function of threonine synthase from yeast. The Journal of biological chemistry. 2002;277:12396-405. doi:10.1074/jbc.M108734200. PMID:11756443
- Omi R, Goto M, Miyahara I, Mizuguchi H, Hayashi H, Kagamiyama H, Hirotsu K. Crystal structures of threonine synthase from Thermus thermophilus HB8: conformational change, substrate recognition, and mechanism. The Journal of biological chemistry. 2003;278:46035-45. doi:10.1074/jbc.M308065200. PMID:12952961
- Thomazeau K, Curien G, Dumas R, Biou V. Crystal structure of threonine synthase from Arabidopsis thaliana. Protein science: a publication of the Protein Society. 2001;10:638-48. doi:10.1110/ps.44301. PMID:11344332
- Mas-Droux C, Biou V, Dumas R. Allosteric threonine synthase. Reorganization of the pyridoxal phosphate site upon asymmetric activation through S-adenosylmethionine binding to a novel site. The Journal of biological chemistry. 2006;281:5188-96
- Baugh L, Gallagher LA, Patrapuvich R, Clifton MC, Gardberg AS, Edwards TE, Armour B, Begley DW, Dieterich SH, Dranow DM, et al. Combining functional and structural genomics to sample the essential Burkholderia structome. PloS one 2013;8:e53851. doi:10.1371/journal.pone.0053851. PMID:23382856
- Covarrubias AS, Hogbom M, Bergfors T, Carroll P, Mannerstedt K, Oscarson S, Parish T, Jones TA, Mowbray SL. Structural, biochemical, and in vivo investigations of the threonine synthase from Mycobacterium tuberculosis. Journal of molecular biology. 2008;381:622-33. doi:10.1016/j.jmb.2008.05.086. PMID:18621388
- Murakawa T, Machida Y, Hayashi H. Product-assisted catalysis as the basis of the reaction specificity of threonine synthase. The Journal of biological chemistry. 2011;286:2774-84. doi:10.1074/jbc.M110.186205. PMID:21084312
- Curien G, Biou V, Mas-Droux C, Robert-Genthon M, Ferrer JL, Dumas R. Amino acid biosynthesis: new architectures in allosteric enzymes. Plant physiology and biochemistry: PPB / Societe francaise de physiologie vegetale. 2008;46:325-39. doi:10.1016/j.plaphy.2007.12.006
- Curien G, Job D, Douce R, Dumas R. Allosteric activation of Arabidopsis threonine synthase by S-adenosylmethionine. Biochemistry. 1998;37:13212-21. doi:10.1021/bi980068f. PMID:9748328
- Madison JT, Thompson JF. Threonine synthetase from higher plants: stimulation by S-adenosylmethionine and inhibition by cysteine. Biochemical and biophysical research communications. 1976;71:684-91. doi:10.1016/0006-291X(76)90842-1. PMID:962947
- Ereño-Orbea J, Majtan T, Oyenarte I, Kraus JP, Martínez-Cruz LA. Structural insight into the molecular mechanism of allosteric activation of human cystathionine β-synthase by S-adenosylmethionine. Proceedings of the National Academy of Sciences. 2014;111:E3845-E52. doi:10.1073/pnas.1414545111
- Sillitoe I, Lewis TE, Cuff A, Das S, Ashford P, Dawson NL, Furnham N, Laskowski RA, Lee D, Lees JG, et al. CATH: comprehensive structural and functional annotations for genome sequences. Nucleic acids research. 2015;43:D376-D81. doi:10.1093/nar/gku947. PMID:25348408
- Curien G, Dumas R, Ravanel S, Douce R. Characterization of an Arabidopsis thaliana cDNA encoding an S-adenosylmethionine-sensitive threonine synthase. Threonine synthase from higher plants. FEBS letters. 1996;390:85-90
- Krishna SS, Majumdar I, Grishin NV. Structural classification of zinc fingers: survey and summary. Nucleic acids research. 2003;31:532-50. doi:10.1093/nar/gkg161. PMID:12527760
- Kaur G, Subramanian S. The insertion domain 1 of class IIA dimeric glycyl-tRNA synthetase is a rubredoxin-like zinc ribbon. Journal of structural biology. 2015;190:38-46. doi:10.1016/j.jsb.2015.02.004. PMID:25721219
- Kaur G, Subramanian S. The Ku-Mar zinc finger: A segment-swapped zinc ribbon in MarR-like transcription regulators related to the Ku bridge. Journal of Structural Biology. 2015;191:281-9.
- Holm L, Laakso LM. Dali server update. Nucleic Acids Research. 2016; 44:W351-5.
- Wiederstein M, Gruber M, Frank K, Melo F, Sippl MJ. Structure-based characterization of multiprotein complexes. Structure. 2014;22:1063-70. doi:10.1016/j.str.2014.05.005. PMID:24954616
- Aravind L, Koonin EV. DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic acids research. 1999;27:4658-70. doi:10.1093/nar/27.23.4658. PMID:10556324
- Graham DE, Taylor SM, Wolf RZ, Namboori SC. Convergent evolution of coenzyme M biosynthesis in the Methanosarcinales: cysteate synthase evolved from an ancestral threonine synthase. The Biochemical journal. 2009;424:467-78. doi:10.1042/BJ20090999. PMID:19761441
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215:403-10. doi:10.1016/S0022-2836(05)80360-2. PMID:2231712
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25:3389-402. doi:10.1093/nar/25.17.3389. PMID:9254694
- Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A. FFAS03: a server for profile–profile sequence alignments. Nucleic acids research. 2005;33:W284-8. doi:10.1093/nar/gki418. PMID:15980471
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR. HMMER web server: 2015 update. Nucleic acids research. 2015;43:W30-8. doi:10.1093/nar/gkv397
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic acids research. 2005;33:W244-8. doi:10.1093/nar/gki408. PMID:15980461
- Christen P, Mehta PK. From cofactor to enzymes. The molecular evolution of pyridoxal-5′-phosphate-dependent enzymes. Chemical record (New York, NY). 2001;1:436-47
- Donini S, Percudani R, Credali A, Montanini B, Sartori A, Peracchi A. A threonine synthase homolog from a mammalian genome. Biochemical and biophysical research communications. 2006;350:922-8. doi:10.1016/j.bbrc.2006.09.112. PMID:17034760
- Harde C, Neff K-H, Nordhoff E, Gerbling K-P, Laber B, Pohlenz H-D. Syntheses of homoserine phosphate analogs as potential inhibitors of bacterial threonine synthase. Bioorganic & Medicinal Chemistry Letters. 1994;4:273-8
- Sanders BD, Jackson B, Marmorstein R. Structural basis for sirtuin function: what we know and what we don't. Biochimica et biophysica acta. 2010;1804:1604-16. doi:10.1016/j.bbapap.2009.09.009. PMID:19766737
- Chen JM, Hou C, Wang G, Tsodikov OV, Rohr J. Structural insight into MtmC, a bifunctional ketoreductase-methyltransferase involved in the assembly of the mithramycin trisaccharide chain. Biochemistry. 2015;54:2481-9. doi:10.1021/bi501462g. PMID:25587924.
- Das K, Acton T, Chiang Y, Shih L, Arnold E, Montelione GT. Crystal structure of RlmAI: implications for understanding the 23S rRNA G745/G748-methylation at the macrolide antibiotic-binding site. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4041-6. doi:10.1073/pnas.0400189101. PMID:14999102.
- Berry MB, Phillips GN, Jr. Crystal structures of Bacillus stearothermophilus adenylate kinase with bound Ap5A, Mg2+ Ap5A, and Mn2+ Ap5A reveal an intermediate lid position and six coordinate octahedral geometry for bound Mg2+ and Mn2+. Proteins. 1998;32:276-88. doi:10.1002/(SICI)1097-0134(19980815)32:3<276::AID-PROT3>3.0.CO;2-G. PMID:9715904.
- Bruender NA, Thoden JB, Kaur M, Avey MK, Holden HM. Molecular architecture of a C-3′-methyltransferase involved in the biosynthesis of D-tetronitrose. Biochemistry. 2010;49:5891-8. doi:10.1021/bi100782b. PMID:20527922.
- Gophna U, Bapteste E, Doolittle WF, Biran D, Ron EZ. Evolutionary plasticity of methionine biosynthesis. Gene. 2005;355:48-57. doi:10.1016/j.gene.2005.05.028. PMID:16046084.
- Jander G, Joshi V. Aspartate-Derived Amino Acid Biosynthesis in Arabidopsis thaliana. The Arabidopsis book / American Society of Plant Biologists. 2009;7:e0121. doi:10.1199/tab.0121.
- Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell research. 2009;19:271-3. doi:10.1038/cr.2009.6. PMID:19153597.
- TheUniProtConsortium. UniProt: A hub for protein information. Nucleic acids research. 2015;43:D204-12. doi:10.1093/nar/gku989. PMID:25348405.
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Computer applications in the biosciences: CABIOS. 1992;8:275-82. PMID:1633570.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular biology and evolution. 2013;30:2725-9. doi:10.1093/molbev/mst197. PMID:24132122.
- Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Research. 2016;44:W242-5.
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. CDD: NCBI's conserved domain database. Nucleic acids research. 2015;43:D222-6. doi:10.1093/nar/gku1221. PMID:25414356.
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658-9. doi:10.1093/bioinformatics/btl158. PMID:16731699.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004;32:1792-7. doi:10.1093/nar/gkh340. PMID:15034147.